Джеймс Уотсон - ДНК. История генетической революции

- Название:ДНК. История генетической революции
- Автор:
- Жанр:
- Издательство:Издательство Питер
- Год:2018
- Город:Санкт-Петербург
- ISBN:978-5-4461-0549-6
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Джеймс Уотсон - ДНК. История генетической революции краткое содержание
ДНК. История генетической революции - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
В конце 1970-х годов цитогенетики (ученые, изучающие хромосомы под микроскопом) обнаружили аномалию в коротком плече X-хромосомы, встречающейся у тех считаных девочек, которые подвержены миодистрофии Дюшенна. Речь идет об участке Xp21. Анализ сцепления, выполненный Бобом Уильямсоном из Медицинской школы при больнице Святой Марии в Лондоне, а также его коллегой Кей Дэвис (в замужестве Дейм), подтвердил, что все дело в участке Xp21. Тем временем генетики-клиницисты воспользовались полиморфизмами длин рестрикционных фрагментов в качестве диагностического инструмента, позволяющего определить, кто из членов той или иной семьи (не исключая этапа внутриутробного развития) несет такую мутацию. Например, если у женщины есть мутация, провоцирующая миодистрофию Дюшенна, то ее сын с вероятностью 50 % заболеет этим недугом. Впервые в истории с помощью метода ПДРФ врачам удалось внутриутробно определить риск рождения ребенка с генетическим заболеванием.
Такой метод диагностики впервые представили в 1978 году сотрудники Калифорнийского университета в Сан-Франциско Юэт Кан и его коллеги. Они использовали сцепленные ПДРФ для диагностики плода с β-талассемией. ДНК для анализа забиралась либо из амниотической жидкости, в которой присутствуют клетки плода, либо из плаценты. Такая процедура называется «биопсия хориона». Правда, точность этого метода не составляла 100 %, поскольку, если вы помните из первой главы, при возникновении яйцеклеток хромосомы подвергаются рекомбинации. Если такая перестановка генетического материала происходит между ПДРФ и интересующим нас геном, то результат биопсии получится неверным, отсюда и погрешность. Она составляет при раннем обнаружении миодистрофии Дюшенна около 5 % и является неизбежным следствием рекомбинации. Но результаты пренатальной диагностики целиком и полностью зависели от выявления конкретного гена, а не от маячковых маркеров.
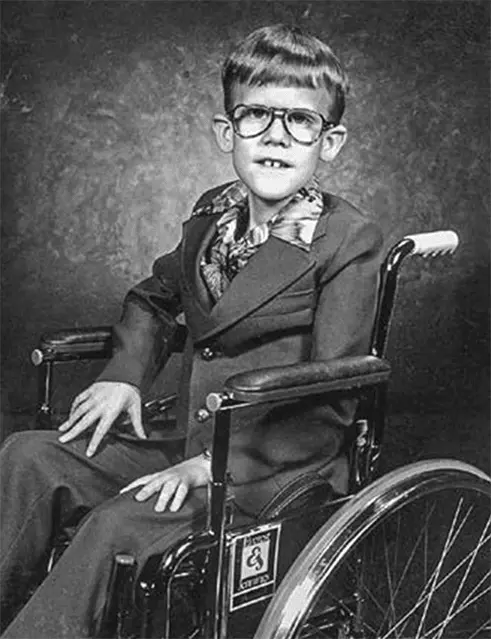
Брюс Брайер, чья частичная делеция X-хромосомы позволила идентифицировать ген миодистрофии Дюшенна. Брюс погиб в автокатастрофе в возрасте семнадцати лет, но прожил относительно полноценную жизнь и даже был отличным органистом.
Ген миодистрофии Дюшенна удалось идентифицировать благодаря мальчику по имени Брюс Брайер, у которого в коротком плече X-хромосомы не хватало очень большого фрагмента. Этот участок был таким большим, что Брюс страдал не только миодистрофией Дюшенна, но и еще тремя другими генетическими заболеваниями. В 1985 году Лу Кункель из Гарвардской медицинской школы рассудил, что мог бы воспользоваться ДНК Брюса, чтобы выделить нормальный ген из ДНК здорового мальчика. Кункель осознал, что вся ДНК Брюса должна быть и у здорового мальчика, а те фрагменты, которые есть у здорового мальчика, но отсутствуют у Брюса, и несут разгадку. Воспользовавшись рекомбинантной технологией, Лу Кункель извлек из нормальной ДНК ту часть, которая была у Брюса, и стал изучать отличающиеся фрагменты, по его мнению, именно в них должен был находиться ген миодистрофии Дюшенна.
Полностью тайна заболевания была разгадана Тони Монако, аспирантом Лу Кункеля. Оказалось, что последовательность pERT87 отсутствует у пятерых мальчиков, страдающих миодистрофией Дюшенна; она располагалась очень близко к искомому гену, иногда даже входила в его состав. Теперь гену уже можно было дать подходящее название: дистрофин. Несколько лет этот ген считался самым крупным в геноме человека, поскольку в нем очень много больших интронов, но затем дистрофин уступил первенство гену, кодирующему другой мышечный белок и не менее удачно названному «титин» [17] Строчка «выделил ген наследственной болезни» обычно хорошо смотрится в резюме; ныне Монако возглавляет Университет Тафта в Бостоне.
.
Эти новые знания были немедленно использованы для пренатальной диагностики миодистрофии Дюшенна. Однако, несмотря на то что функция дистрофина была исследована на протяжении десятилетий, мы по-прежнему не в силах эффективно лечить или облегчать течение миодистрофии Дюшенна. Именно это удручает в текущем положении дел: генетика позволяет идентифицировать и понять болезнь, но пока в большинстве случаев не позволяет исправить генетические ошибки. Подобные истории связаны и с болезнью Хантингтона, и с муковисцидозом, и с другими наиболее серьезными генетическими расстройствами. По сравнению с разработкой препарата, позволяющего блокировать известную мишень, лечение генетического заболевания – вызов совершенно иного плана: здесь требуется добавить недостающие гены или заменить нефункциональные. В случае с миодистрофией Дюшенна речь идет как раз о том, как доставлять достаточно крупный белок к мышцам.
Среди некоторых наиболее перспективных методов борьбы с миодистрофией Дюшенна рассматривается возврат к методу генетической терапии, который ранее считался совершенно безнадежным, об этом мы поговорим далее в главе. Исследователи пробуют организовать доставку дистрофина в мышечные волокна при помощи вирусов – речь идет либо о полноценном гене, либо о его уменьшенной версии. Другой метод больше подходит тем немногим пациентам с миодистрофией Дюшенна, которые страдают от иной мутации: она преждевременно блокирует трансляцию транспортной РНК дистрофина. Биотехнологические компании, например PTC Therapeutics , разрабатывают препараты, позволяющие рибосоме миновать ложные стоп-сигналы для получения полноценной коммуникации, на основе которой выстраивается полноценный белок. В 2016 году компания Sarepta Therapeutics получила одобрение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) на препарат этеплирсен, увы, это лекарство предназначено для лечения немногих пациентов, а его клиническая эффективность остается до конца невыясненной. Наконец, Кей Дэвис и ее коллеги из Оксфордского университета разработали метод замены недостающего дистрофина, искусственно усиливая экспрессию родственного гена, который называется утрофин. Этот метод хорошо работает на мышиных моделях с миодистрофией Дюшенна, пока ведутся клинические испытания.
Наиболее интенсивные поиски генов наследственных болезней пришлись на 1980-е годы, когда врачи пытались побороть одно из самых распространенных генетических заболеваний – муковисцидоз. Охота за геном муковисцидоза вышла особенно примечательной по двум причинам: впервые коммерческая компания участвовала в картировании гена человеческой болезни, и впервые развернулось жестокое соперничество среди ученых, причастных к этому направлению исследований.
Читать дальшеИнтервал:
Закладка:









