Алексей Яковлев - Реабилитация при болезни Паркинсона

- Название:Реабилитация при болезни Паркинсона
- Автор:
- Жанр:
- Издательство:неизвестно
- Год:неизвестен
- ISBN:9785449682420
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Алексей Яковлев - Реабилитация при болезни Паркинсона краткое содержание
Реабилитация при болезни Паркинсона - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
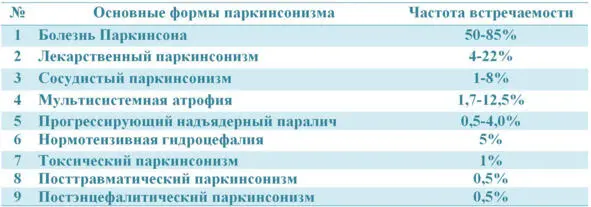
Таб. 1. Частота основных форм паркинсонизма.
В основе патогенеза развития БП лежит повреждение дофаминергических нигростриарных нейронов и накопление альфа-синуклина (αSyn), содержащихся в тельцах Леви. Альфа-синуклеин является основным компонентом телец Леви. Нарушение процессинга αSyn является центральным звеном молекулярного патогенетического каскада, ведущего к накоплению в клетке нерастворимых белковых комплексов и прогрессирующей дегенерации соответствующей популяции нейронов при БП. Еще в 1919 году советский невропатолог Константин Николаевич Третьяков (рис. 3) связал развитие симптомов паркинсонизма с утратой пигментных нейронов черной субстанции ствола, а также с накоплением в этих клетках патологических включений (рис. 4). Нейрональные включения К. Н. Третьяков предложил называть тельцами Леви в честь немецкого морфолога Фридриха Леви, который ранее описал схожие изменения при паркинсонизме в других отделах ствола мозга.

Рис. 3. Основоположник нигральной теории паркинсонизма, профессор К. Н. Третьяков.
Накопление αSyn рассматривается как основной момент в патогенезе БП, что отражено в полиморфизмах гена SNCA, ведущих к образованию аномального белка. Убедительным доказательством нейротоксичности αSyn стало получение на основе гиперэкспресcии гена SNCA человека трансгенных животных (дрозофила, мышь), демонстрирующих нейрональные αSyn-положительные включения и возрастную нейродегенерацию дофаминергических нейронов мозга.
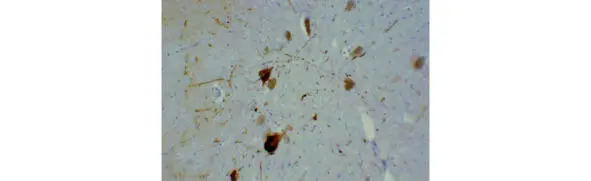
Рис. 4. Тельца Леви в черной субстанции среднего мозга под микроскопом в 20-кратном увеличении (Suraj Rajan).
Стоит заметить, что БП является преимущественно спорадическим заболеванием (90—95% случаев), семейный анамнез прослеживается всего лишь в 5—15% случаев по данным различных источников.
В настоящее время доказано, что наследственные варианты БП обусловлены мутациями генов SNCA, LRRK2, PRKN, DJ1, PINK1 и ATP13A2 и др. (рис. 5)
Мутация гена, расположенного на хромосоме 4q21—22, кодирующего белок αSyn, обуславливает развитие наследственной формы БП с аутосомно-доминантным типом наследования. Мультипликация генного локуса PARK1 увеличивает экспрессию αSyn и вызывает БП. Таким образом, можно утверждать, что повышение экспрессии αSyn токсично для нейронов.
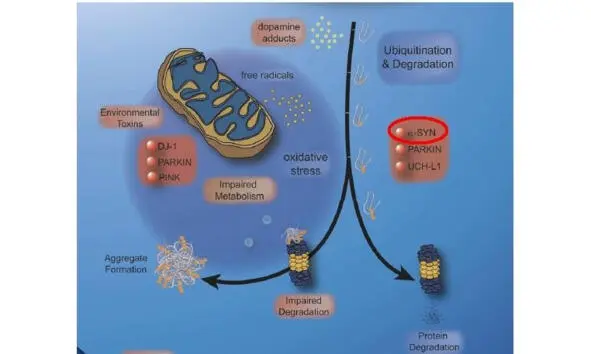
Рис. 5. Мутации пяти генов являются причиной развития ювенильной формы БП, включая мутацию гена αSyn.
Также показано ингибирующее влияние αSyn на процесс мембранного слияния, который является важным биологическим механизмом поддержания базовой клеточной организации у эукариот. F. Kamp и соавт. (2010) на культуре клеток Caenorhabditis elegans (свободноживущая нематода) показали, что повышенная концентрация αSyn приводит к митохондриальной фрагментации, а также может смещать динамическое морфологическое равновесие митохондрий к уменьшенному слиянию. В свою очередь, митохондриальная фрагментация, вызванная экспрессией αSyn, возобновляется коэкспрессией PINK1, паркина или DJ-1, но не БП-ассоциированными мутациями гена PINK1 G309D, гена паркина Δ1—79 или гена DJ-1 C106A. Мутации в гене альфа-синуклеина (А53Т, А30Р) сопровождаются нарушением стабильности центральной части белковой молекулы, изменением её пространственной организации и формированием бета-складчатых слоев, способных аггрегировать с другими аналогичными молекулами с образованием мультимолекулярных фибрилл, что также нарушает процессы физиологического мембранного слияния.
Мутации генного локуса PARK2 связаны с развитием ювенильной БП с аутосомно-рецессивным типом наследования, болезни Альцгеймера, рака, сахарного диабета. В 1998 году был идентифицирован основной ген аутосомно-рецессивной ювенильной БП в хромосомной области 6q25.2—27, содержащий 12 экзонов, и кодирующий белок паркин, локализованный в комплексе Гольджи и цитозоле нейронов подкорковых ядер головного мозга. Наибольшая концентрация паркина обнаружена в пигментных клетках компактной зоны черной субстанции. Паркин обладает свойствами убиквитин-лигазы и играет ключевую роль в клеточной деградации аномальных белков. Мутации в гене паркина ведут к нарушению функций данного фермента в черной субстанции и стриатуме, что сопровождается накоплением аномальных белковых субстратов в клетке, индукцией апоптоза и гибелью нейронов.
В российском исследовании (Иллариошкин С. Н. и соавт., 2007 г.) с клинико-генетическим анализом 26 больных из 20 семей с возрастом дебюта БП до 30 лет были получены следующие данные: 41% семей имели мутацию гена паркина (PRKN). Всего было выявлено 9 мутаций: 6 из них были представлены делецией отдельных экзонов, 3-точковыми мутациями гена PRKN, ведущими к сдвигу рамки считывания (del202—203AG) или нарушению сплайсинга (IVS1+1G/А). T. Yoshida и соавт. (2010) на примере мутантных мышей (mnd2) показали, что введение таким мышам нормального белка паркина не влияет на течение нейродегенеративного процесса при БП.
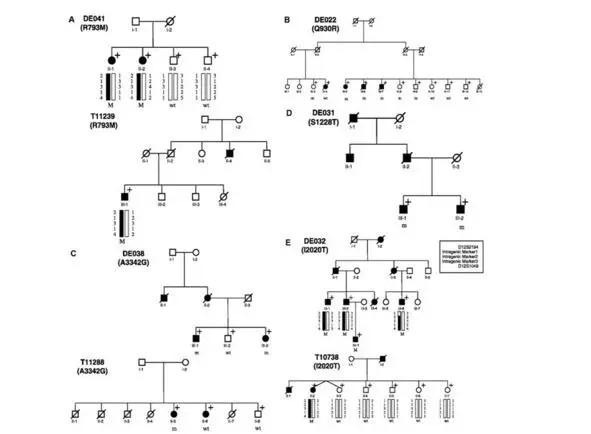
Рис. 6. Родословные семей с мутациями гена LRRK2. Закрашенные символы – члены семей с клиническими проявлениями БП. Знаком «+» обозначены члены семей, которым проведено молекулярно-гентическое тестирование: «m» (mutation) – обозначены носители мутации гена LRRK2, «wt» (wild type) – носители нормального гена LRRK2 (Berg D. et al., 2005).
Наиболее распространенной причиной моногенной формы БП с аутосомно-доминантным типом наследования является мутация гена LRRK2 (leucinerich repeat kinase 2), картированная на хромосоме 12q12, сцепленная с локусом PARK8 и кодирующая белок дардарин, функции которого изучены недостаточно (рис. 6). По некоторым данным, дардарин принимает участие в процессинге нейрональных белков, функционировании митохондрий и в межнейронных взаимодействиях. J. Vitte и соавт. (2010) исследовали экспрессию этого белка в мозге здоровых людей и людей, страдающих БП. Установлено, что эндогенный белок LRRK2 локализован в эндоплазматическом ретикулуме. До 24% белка обнаружено в клеточном ядре, в частности в 11% случаев – в тельцах Леви черной субстанции головного мозга. У пациентов, имеющих мутацию гена LRRK2 на хромосоме 12q12, концентрация исследуемого белка была увеличена до 50%. H. Mortiboys и соавт. (2010) провели исследование биоптатов кожи пяти пациентов, имеющих мутацию гена LRRK2 (G2019S), с целью изучения митохондриальных потенциалов, уровня внутриклеточной аденозин-трифосфатазы и продукции АТФ митохондриями нейронов. В результате исследования выявлена митохондриальная элонгация, снижение уровня продукции внутриклеточной АТФ, а также снижение мембранных потенциалов митохондрий.
Читать дальшеИнтервал:
Закладка:









