Эрик Кандель - Расстроенная психика. Что рассказывает о нас необычный мозг

- Название:Расстроенная психика. Что рассказывает о нас необычный мозг
- Автор:
- Жанр:
- Издательство:Литагент Corpus
- Год:2021
- Город:М.
- ISBN:978-5-17-119013-2
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Эрик Кандель - Расстроенная психика. Что рассказывает о нас необычный мозг краткое содержание
В формате PDF A4 сохранен издательский макет книги.
Расстроенная психика. Что рассказывает о нас необычный мозг - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Роль белков в болезни Альцгеймера
Что же приводит к формированию бляшек и клубков? Ученые установили, что в образовании амилоидных бляшек повинен бета-амилоидный пептид . Этот пептид входит в состав более крупного белка, называемого предшественником бета-амилоида (APP), который, как считается, заякорен в клеточной мембране дендритов – коротких ветвистых отростков нейронов (рис. 5.7). Два разных фермента разрезают белок-предшественник в двух разных местах, высвобождая тем самым амилоидный пептид. Покинув клеточную мембрану, пептид обосновывается в пространстве возле нейрона.
Как выяснилось, синтез и высвобождение бета-амилоидного пептида – совершенно нормальные события для мозга каждого человека. Однако у людей, страдающих болезнью Альцгеймера, может ускоряться производство белка или замедляться очистка от него околоклеточного пространства. И то, и другое может приводить к аномальному накоплению этих пептидов, которые вдобавок довольно “липкие”. Они слипаются друг с другом, в конце концов формируя характерные для болезни Альцгеймера амилоидные бляшки.
Другой белок, вовлеченный в развитие болезни Альцгеймера, называется тау и содержится внутри нейрона. Для нормальной работы этот белок должен приобретать трехмерную форму. Он принимает такую форму в результате фолдинга – процесса, при котором аминокислотная последовательность, составляющая белок, сворачивается в специфическую конфигурацию. Это можно представить как невероятно сложное оригами. Если какой-то молекулярный дефект приводит к неправильному сворачиванию тау-белка, формируются токсичные сгустки (рис. 5.8), собирающиеся в нейрофибриллярные клубки.
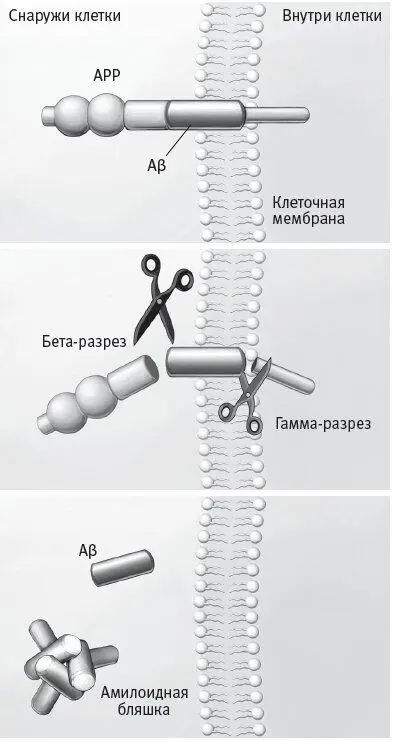
Рис. 5.7.Схема образования амилоидной бляшки. Предшественник бета-амилоида (APP), находящийся в клеточной мембране, содержит бета-амилоидный (Aβ) пептид ( вверху ). Два фермента разрезают предшественник бета-амилоида: сначала происходит бета-разрез, затем гамма-разрез ( посередине ). Эти разрезы высвобождают бета-амилоид в пространство возле клетки, где он может образовывать амилоидные бляшки ( внизу ).
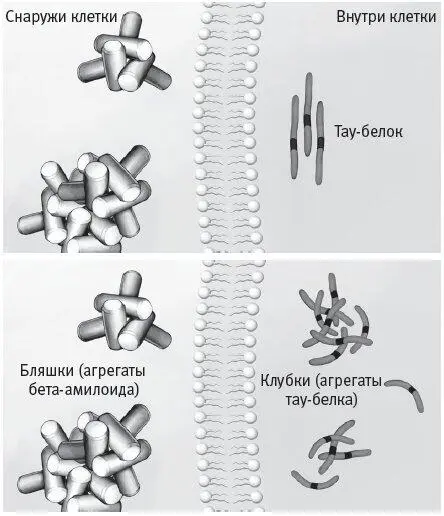
Рис. 5.8.Схема образования нейрофибриллярного клубка. Молекулярный дефект приводит к неправильному фолдингу тау-белка. Когда такое происходит, белок скапливается внутри клетки, формируя нейрофибриллярные клубки.
В сочетании два типа агрегатов – бляшки снаружи нервной клетки и клубки внутри – приводят к гибели нейронов и прогрессированию болезни Альцгеймера.
Генетические исследования болезни Альцгеймера
Хотя болезнь Альцгеймера обычно поражает людей старше 70 или 80, у которых не было родственников с тем же диагнозом, редкие формы болезни с ранним началом передаются по наследству. Джону Харди из Лондонского университетского колледжа представилась необычная возможность изучить генетическую основу болезни Альцгеймера, когда с ним связалась Кэрол Дженнингс.
В начале 1980-х у 58-летнего отца Кэрол обнаружили болезнь Альцгеймера. Вскоре болезнь развилась у его сестры и брата, которым тоже еще не было 60. Оказалось, что той же болезнью страдали прадед Кэрол, дед и двоюродный дед. В основной ветви семьи болезнь Альцгеймера развилась у 5 из 10 детей, причем в одно и то же время. Средний возраст начала болезни составлял около 55 лет (вообще, самое раннее начало наследственной формы болезни Альцгеймера зафиксировали у человека, которому не было и 30).
Харди с коллегами решил выяснить, какие генетические особенности унаследовали все заболевшие сиблинги в семье Дженнингс, но не унаследовал ни один из незаболевших. Они установили, что пять сиблингов и один кузен, у которых развилась болезнь Альцгеймера, обладали идентичным фрагментом хромосомы 21, самой маленькой в геноме человека. Однако у двух здоровых сиблингов тоже нашли небольшой идентичный участок в том же фрагменте. На основании этого Харди сделал вывод, что ген, ответственный за болезнь Альцгеймера, находится не в том участке 21-й хромосомы, который был одинаковым и у больных, и у здоровых сиблингов. Затем он внимательно изучил ту часть фрагмента 21-й хромосомы, по которой больные сиблинги отличались от здоровых, и нашел дефектный ген, ответственный за агрегацию бета-амилоида 55 .
Так был выявлен первый ген, участвующий в развитии болезни Альцгеймера, и положено начало ее исследованию. Патологи уже поняли, что бета-амилоидный пептид формирует бляшки, но Харди показал, что в семье Дженнингс болезнь развивается из-за мутации гена предшественника бета-амилоида, заставляющей пептидные молекулы слипаться.
Впоследствии Харди и другие ученые обнаружили множество других мутаций. Научный коллектив из Торонто нашел семьи с наследственной болезнью Альцгеймера, в которых встречались мутации генов [67] У человека гены PSEN1 (на хромосоме 14) и PSEN2 (на хромосоме 1) кодируют пресенилины 1 и 2 соответственно. Функций у этого мембранного белка немало, и его аномалии могут способствовать нейродегенерации разными путями. В частности, он представляет собой “режущий” компонент гаммасекретазы – ферментного комплекса, делающего гамма-разрез предшественника бета-амилоида. Четко фиксированного места разреза нет, и молекулы бета-амилоида получаются немного разными по длине. Мутации гена пресенилина смещают соотношение этих форм в пользу длинной (недорезанной), более склонной к агрегации.
, кодирующих белок пресенилин 56 . Мутации не позволяют пресенилину нормально участвовать в вырезании бета-амилоида для его выхода в межнейронное пространство. Эти результаты прекрасно согласуются с находкой Харди. Оба исследования показывают, что все семьи, в которых отмечается раннее начало болезни Альцгеймера, несут мутации, способствующие формированию опасных бета-амилоидных бляшек в мозге. Иными словами, все мутации сходятся на одном пути, который ведет к развитию наследственной формы болезни Альцгеймера с ранним началом (рис. 5.9).
Эти генетические исследования семей с наследственной болезнью Альцгеймера побудили ученых выяснить, а нет ли мутаций, снижающих количество бета-амилоида? И если такие мутации есть, то защищают ли они от болезни Альцгеймера?
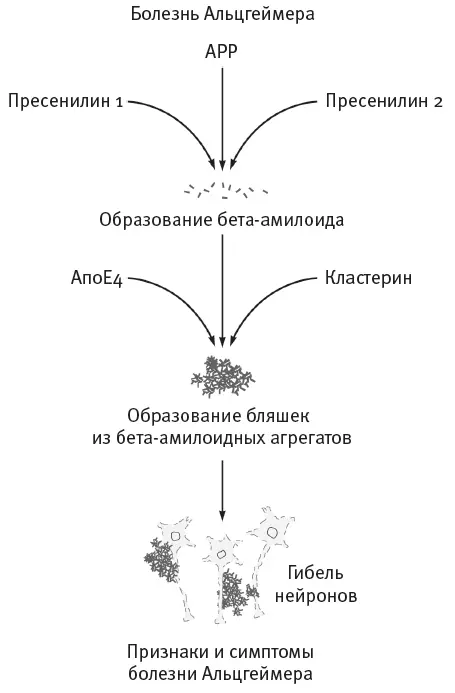
Рис. 5.9.Несколько разных путей, ведущих к развитию болезни Альцгеймера с ранним началом, вливаются в магистраль производства одного продукта – бета-амилоидных скоплений. Для людей с болезнью Альцгеймера характерно избыточное производство белка кластерина . Он взаимодействует с бета-амилоидом, усугубляя потерю нервной ткани в энторинальной коре. APP – предшественник бета-амилоида. АпоЕ4 – неблагоприятная изоформа аполипопротеина Е.
Читать дальшеИнтервал:
Закладка:








