Коллектив авторов - Клиническая патофизиология

- Название:Клиническая патофизиология
- Автор:
- Жанр:
- Издательство:Литагент СпецЛит
- Год:неизвестен
- ISBN:978-5-299-00511-0
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Коллектив авторов - Клиническая патофизиология краткое содержание
Данные лекции базируются на основе учебной программы по дисциплине «Патофизиология» и предназначены для интернов, клинических ординаторов, аспирантов (адъюнктов), преподавателей курсов повышения квалификации, врачей, обучающихся в системе послевузовского профессионального образования, а также для студентов старших курсов медицинских вузов (курсантов).
Клиническая патофизиология - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
К неспецифическим механизмам относят расстройства энергетического обеспечения; повреждение мембран и ферментов клеток; дисбаланс ионов и воды в них; нарушения в геноме и/или механизмов экспрессии генов нейронов; расстройства регуляции функций клеток.
К специфическим механизмам повреждения при ишемических повреждениях головного мозга относят звенья «ишемического каскада» (рис. 3.2).
Основными звеньями «ишемического каскада» при цереброваскулярных заболеваниях являются следующие:
– снижение мозгового кровотока (энергетический дефицит);
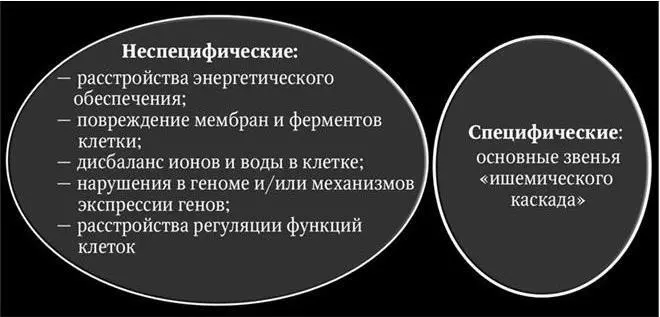
Рис. 3.2 . Механизмы повреждения нейронов при цереброваскулярных заболеваниях
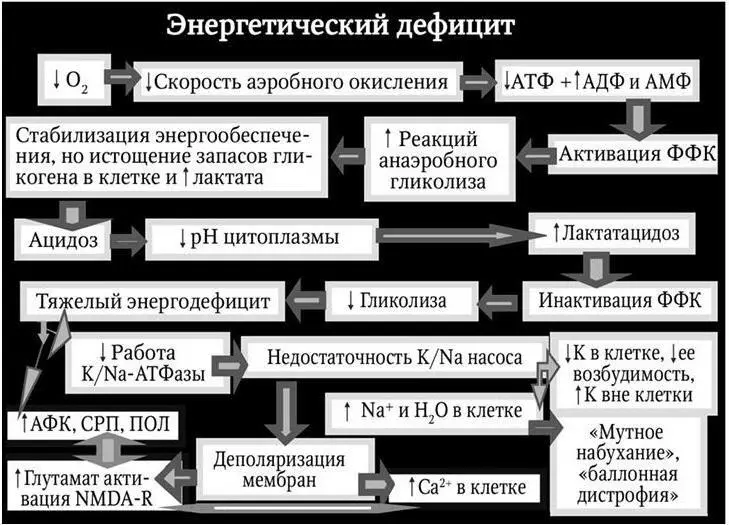
Рис. 3.3. Начальный этап «ишемического каскада» при гипоксии
– избыток глутамата и глутаматная «эксайтотоксичность»;
– внутриклеточное накопление избытка кальция;
– активация внутриклеточных ферментов;
– повышение генерации активных форм кислорода (АФК), активация свободнорадикальных процессов (СРП) с развитием «оксидативного» или «оксидантного стресса»;
– экспрессия генов раннего реагирования;
– отдаленные последствия ишемии (реакции местного воспаления, микроваскулярные нарушения, повреждения гематоэнцефалического барьера и др.);
– некроз, апоптоз нейронов.
На начальном этапе «ишемического каскада» (рис. 3.3) при кислородном голодании любой этиологии в митохондриях снижается скорость аэробного окисления и окислительного фосфорилирования. Это ведет к понижению количества аденозинтрифосфата (АТФ) и возрастанию содержания АДФ и АМФ (аденозинди- и аденозинмонофосфата). Уменьшается коэффициент АТФ/АДФ + + АМФ, снижаются функциональные возможности клетки. При низком соотношении АТФ/АДФ + АМФ активируется фермент фосфофруктокиназа (ФФК). Это позволяет увеличить интенсивность реакций анаэробного гликолиза. Клетка расходует гликоген, обеспечивая себя энергией за счет бескислородного метаболизма глюкозы. В результате этого происходит относительная адаптация нейронов к гипоксии и энергообеспечение стабилизируется. Однако это сопровождается истощением запасов гликогена в клетке. Морфологически на этой стадии процесса в клетках отмечается исчезновение гликогеновых гранул. На системном уровне в организме гипоксия провоцирует стресс и гормоны стресса – катехоламины и глюкокортикоиды, которые усиливают гликолиз, гликогенолиз, глюконеогенез и транспорт экзогенной глюкозы в наиболее жизненно важные органы и ткани.
Цена такой адаптации высока не только вследствие меньшей энергетической эффективности анаэробного пути по сравнению с аэробным. Конечным продуктом гликолиза является молочная кислота. Если кислород доступен, то лактат окисляется полностью. Острая гипоксия создает условия для значительного возрастания содержания лактата и свободного фосфата в клетках и в крови. Это вызывает внутриклеточный ацидоз. Считается, что ацидоз сам по себе не разрушает клеточных мембран. Более того, его можно рассматривать в качестве защитной реакции клетки, так как умеренное снижение pH оказывает стабилизирующее действие на клеточные мембраны. Однако прогрессирующий ацидоз может вызвать денатурацию некоторых внутриклеточных белков и формирование в цитоплазме включений («мутное набухание», «зернистая дистрофия»). Наибольшее патогенное значение для клетки имеет подавление в условиях ацидоза активности ФФК. В зрелых клетках ФФК «кислотоугнетаемый» фермент. Гипоксия активирует гликолиз, ведущий к ацидозу, ацидоз тормозит реакции гликолиза. В этой стадии гипоксии в клетке формируется дефицит АТФ, поскольку эффективность аэробного механизма снижена из-за кислородного дефицита, а анаэробного – в связи с ацидозом.
В наибольшей мере повреждаются основные клеточные потребители энергии: градиентсоздающие и сократительные механизмы клетки. Весьма энергоемким ферментом является мембранная калий-натриевая аденозинтрифосфотаза (АТФаза). Дефицит энергии приводит к утрате калий-натриевого градиента. Это порождает несколько последствий. Клетки утрачивают ионы калия, а вне клеток возникает их избыток. Частичная утрата потенциала покоя делает клетки менее возбудимыми.
Недостаточность калий-натриевого насоса, сопряженная с деполяризацией мембран, приводит к ряду негативных последствий (см. рис. 3.3): накоплению избытка натрия и жидкости в клетке; активации глутаматных рецепторов N-метил-D-аспартата (NMDA); накоплению избытка кальция в клетке. Важнейшим из прямых последствий повреждения калий-натриевого насоса является накопление избытка натрия в клетке. Из-за осмотической активности натрия развивается гипергидратация клеток. На этой стадии прогрессирующей гипоксии выявляются морфологические признаки «мутного набухания» и «баллонной дистрофии» нейронов.
Следующий этап «ишемического каскада» включает несколько типовых патогенетических механизмов. К числу наиболее значимых из этих механизмов относят: глутаматную эксайтотоксичность; кальциевый, «оксидативный стресс»; липопероксидный; избыточный респираторный взрыв.
Важным механизмом повреждения нейронов является чрезмерная продукция нейронами нейротрансмиттерных аминокислот (глутамата и аспартата) и выделение их из аксонов, что наблюдается уже в течение первых 10 – 30 мин с момента острой локальной ишемии. Установлено, что концентрация глутамата может вернуться к исходному уровню при условии восстановления кровотока через 30 – 40 мин от начала острой ишемии (глутамат – основной возбуждающий нейротрансмиттер ЦНС. Он вовлечен в большое число нейрональных и глиальных процессов, участвует в формировании высших когнитивных механизмов; в высоких концентрациях он нейротоксичен). В периишемической зоне мозга нейроны и клетки нейроглии поглощают глутамат из межклеточного пространства. У клеток же ишемизированной области мозга для этого недостаточно энергии. Снижение обратного захвата глутамата и аспартата астроглией приводит к перевозбуждению ионотропных рецепторов NMDA-рецептеров, метаболотропных и AMPA-рецепторов, регулирующих уровни K +,Na +,Са 2+,Cl –во внеи внутриклеточном пространстве. Это обуславливает раскрытие контролируемых ими кальциевых каналов и приводит к дополнительному притоку ионов Са 2+в нейроны из интерстиция, а также к высвобождению внутриклеточного Са 2+из депо.
При исследовании механизмов ишемического некробиоза нейронов подтверждена концепция о ключевой роли избытка ионизированного внутриклеточного кальция (как последующего этапа «ишемического каскада») в этом процессе, особенно на его глубоких стадиях. Кальций в данном случае выступает не просто как электролит, а как мощный модулятор клеточных функций, избыток которого токсичен для клетки. Внутриклеточная концентрация кальция поддерживается на уровне 10 – 7М, что в 10 000 раз меньше, чем в межклеточной жидкости. При функционировании здоровых клеток прием внешних химических или электрических сигналов сопровождается кратковременным обратимым повышением внутриклеточной концентрации кальция, что необходимо для ответа на стимул. Кальций проникает внутрь через потенциал-зависимые входные кальциевые каналы. Кроме того, раздражение кальций-мобилизующих рецепторов ведет к активации фосфолипазы С и продукции липидных внутриклеточных посредников – диацилглицерина и инозитолтрифосфата. Последний взаимодействует с мембранами митохондрий и вызывает выход депонированного там кальция в цитоплазму. Цитоплазматический кальций переходит в активную форму путем взаимодействия со своим белковым внутриклеточным рецептором – кальмодулином. Комплекс кальций – кальмодулин активирует кальмодулинзависимые протеинкиназы, которые, вместе с протеинкиназой С, активизируемой диацилглицерином, регулируют активность клеточных ферментов.
Читать дальшеИнтервал:
Закладка:









