Коллектив авторов - Диффузные болезни соединительной ткани

- Название:Диффузные болезни соединительной ткани
- Автор:
- Жанр:
- Издательство:Литагент СпецЛит
- Год:неизвестен
- ISBN:978-5-299-00351-2
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Коллектив авторов - Диффузные болезни соединительной ткани краткое содержание
Руководство предназначено для ревматологов, терапевтов, врачей общей практики (семейных врачей) и студентов медицинских вузов.
Диффузные болезни соединительной ткани - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Мышечная слабость, распространяющаяся на дыхательные мышцы, включая диафрагму, может быть причиной снижения вентиляционной функции легких. Клинически отмечается частое и поверхностное дыхание, инспираторная одышка, развивается гипостатическая пневмония. Дисфагия с аспирацией жидкости и пищи в легкие обусловливает развитие аспирационной пневмонии. Поражение легких выявляется у 5 – 46 % больных ДМ (ПМ), главным образом в виде интерстициальной пневмонии, фиброзирующего альвеолита и фиброза. Диагностируется на основании клинико-рентгенологических, функциональных и морфологических (биопсия) данных.
Одышка и кашель, хрипы и крепитация наблюдаются при выраженном поражении легких. Легочные функциональные тесты указывают на преимущественно рестриктивный тип нарушений со снижением общей и жизненной емкости легких; гипоксемия характеризуется умеренным снижением диффузионной способности легких.
Патология легких при ДМ (ПМ) имеет сложный генез и варьирует в широких пределах. Выделяют определенные субтипы интерстициального поражения легких, которые следует учитывать при диагностике и лечении ДМ (ПМ):
I. Острый или подострый тип, который характеризуется тяжелой быстропрогрессирующей одышкой и нарастающей гипоксемией уже в первые месяцы заболевания, аналогично синдрому Хаммена – Рича.
II. Хронический тип с медленнопрогрессирующей одышкой.
III. Асимптомный тип, который протекает субклинически, выявляется при рентгенологическом и функциональном исследовании легких.
Первый тип интерстициального поражения легких имеет наихудший прогноз и требует ранней активной терапии (глюкокортикостероидами (ГКС), цитостатиками и др.).
Полимиозит может предшествовать легочной патологии, развиться после поражения легких или возникать одновременно с легочными проявлениями. Выделяется также и «амиопатический» ДМ, который может ассоциироваться с тяжелым быстропрогрессирующим поражением легких.
Легочный фиброз, обусловленный интерстициальным поражением ткани легких, легочным васкулитом и развитием септально-альвеолярного склероза, отмечается у 5 – 10 % больных. Он характеризуется нарастающей инспираторной одышкой, сухим кашлем, крепитирующими хрипами в нижних отделах легких, нарастающей дыхательной недостаточностью.
Необходимо иметь в виду возможность развития опухолевого, чаще метастатического, процесса в легких.
Поражения желудочно-кишечного тракта(ЖКТ) отмечаются нередко и проявляются нарастающей дисфагией, отсутствием аппетита, иногда – болью в животе и гастроэнтероколитом.
Ведущей симптоматикой поражения желудочно-кишечного тракта при ДМ (ПМ) является дисфагия. Дисфагия развивается вследствие снижения контрактильной силы фарингеальных мышц и мышц верхнего отдела пищевода, нарушения перистальтики, слабости мышц мягкого нёба и языка. Это обусловливает поперхивание, нарушение глотания твердой и жидкой пищи, которая может выливаться через нос. Фарингеально-пищеводная дисфагия – важный дифференциально-диагностический признак ДМ (ПМ). Голос приобретает носовой оттенок. Дисфония нередко сочетается с дисфагией и у тяжелобольных иногда переходит в афонию.
В отличие от ССД при ДМ (ПМ) поражаются верхние отделы пищевода и глоточное кольцо, поэтому клиническая и рентгенологическая картины различны. При склеродермии жидкая пища проходит хорошо, не выливается через нос, однако рентгенологические признаки поражения и осложнения склеродермического эзофагита выражены сильнее, чем у больных с ДМ (ПМ).
Тяжелая прогрессирующая дисфагия, когда твердая пища срыгивается, а жидкая выливается через нос, из-за возможности аспирации представляет непосредственную угрозу жизни больного и является прямым показанием к срочной терапии максимальными дозами кортикостероидов.
При вовлечении в процесс пищеводного сфинктера возможно развитие рефлюкс-эзофагита.
Описаны отдельные случаи ДМ (ПМ) с желудочно-кишечными кровотечениями, перфорацией желудка, в основе которых лежат васкулит и некрозы, возникающие в пищеварительном тракте.
Умеренное увеличение печени с изменением функциональных проб наблюдается приблизительно у 1/3 больных; реже выявляется гепатолиенальный и железисто-селезеночный синдромы.
Поражения почекпри ДМ (ПМ) встречаются относительно редко. При остром течении тяжелая персистирующая миоглобинурия может привести к развитию почечной недостаточности. Среди больных ДМ (ПМ) у 41,5 % отмечается транзиторная протеинурия с микрогематурией и цилиндрурией (Мазуров В. И., Беляева И. Б., 2005). При остром течении ДМ (ПМ) у отдельных больных могут наблюдаться развитие гломерулонефрита, сосудистая патология почек с фибриноидными изменениями артериол и тромбозом.
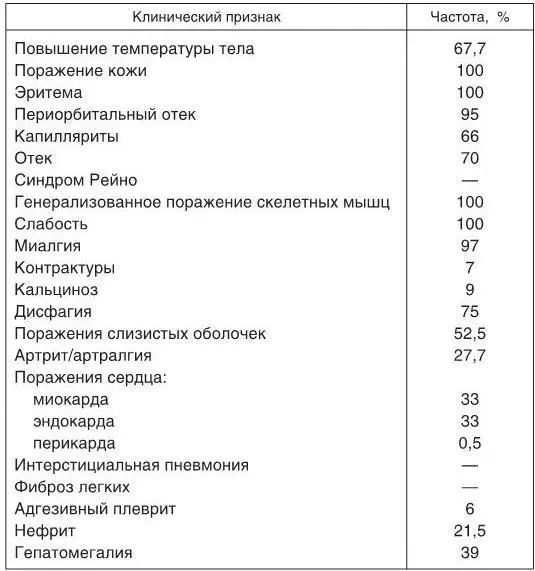
Таблица 1
Частота основных клинических проявлений ДМ (ПМ)
Эндокринные нарушенияпроявляются изменениями функциональной активности половых желез, гипофизарно-надпочечниковой системы, которые могут быть связаны как с тяжестью заболевания и васкулитом, так и с проводимой стероидной терапией.
Поражение нервной системывстречается редко и характеризуется развитием псевдоневрологической симптоматики, хотя у отдельных больных возможно развитие слабо выраженного полиневрита и даже поражений центральной нервной системы (ЦНС) за счет васкулита. Наиболее часто отмечаются вегетативные расстройства. Изредка наблюдаются нарушения психики, эмоциональная неустойчивость больных, которые могут быть связаны с приемом высоких доз ГКС. Частота основных клинических проявлений ДМ (ПМ) по данным Н. Г. Гусевой (2004) представлена в табл. 1.
1.4. КЛИНИКО-ИММУНОЛОГИЧЕСКИЕ ПОДТИПЫ ДЕРМАТОМИОЗИТА
Кроме классических вариантов ДМ и ПМ можно выделить несколько клинико-иммунологических подтипов, развитие которых ассоциируется с синтезом различных типов антител. Эти подтипы различаются не только по спектру клинических проявлений, но и по иммуногенетическим маркерам, прогнозу, ответу на применение ГКС.
Синтез антител к аминоацил-тРНК-синтетазам ассоциируется с развитием так называемого антисинтетазного синдрома, для которого характерны следующие основные признаки: острое начало миозита, интерстициальное поражение легких, лихорадка, симметричный артрит, синдром Рейно, «рука механика», неполный ответ на применение ГКС с частым обострением при снижении дозы препарата, начало заболевания преимущественно в весеннее время. Характерным проявлением антисинтетазного синдрома является интерстициальное поражение легких, которое выявляется у 50 – 70 % больных с наличием антител Jо-1. Развитие артрита чаще наблюдается у больных с наличием антител Jо-1 (57 – 100 %), чем у больных с другими формами миозита. Артрит, как правило, неэрозивный, характеризуется наиболее частым вовлечением в процесс мелких суставов кистей и лучезапястных суставов. Синдром Рейно при антисинтетазном синдроме наблюдается в 60 % случаев.
Читать дальшеИнтервал:
Закладка:









