БСЭ - Большая Советская энциклопедия (ВА)

- Название:Большая Советская энциклопедия (ВА)
- Автор:
- Жанр:
- Издательство:неизвестно
- Год:неизвестен
- ISBN:нет данных
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
БСЭ - Большая Советская энциклопедия (ВА) краткое содержание
Большая Советская энциклопедия (ВА) - читать онлайн бесплатно полную версию (весь текст целиком)
Интервал:
Закладка:
Периодический закон Д. И. Менделеева (1869) вскрыл зависимость В. элемента от его положения в периодической системе (см. Периодическая система элементов Д. И. Менделеева). Элементы одинаковых групп системы обладают одинаковой высшей В., в большинстве случаев равной номеру той группы, в которой находится этот элемент; высшая В. меняется на 1 при переходе от одной группы к соседним. Эта зависимость сыграла чрезвычайно важную роль в развитии химии: зная лишь положение элемента (в том числе элементов, которые в то время ещё не были открыты) в периодической системе, можно было определить его валентные возможности, предсказать состав его соединений и впоследствии синтезировать их. С помощью представлений о формальной (стехиометрической) В. (см. Стехиометрия ) химикам удалось обобщить и систематизировать огромный экспериментальный материал по строению, стехиометрическому составу и свойствам многих десятков и сотен тысяч органических и неорганических соединений.
Первые электронные теории ковалентности и гетеровалентности. До электронных представлений о строении вещества В. трактовалась формально. Лишь в 20 в. было установлено, что химическая связь осуществляется за счёт электронов внешних (валентных) оболочек атомов.
В 1916 Г. Льюис постулировал, что химическая связь осуществляется парой электронов, принадлежащих одновременно обоим взаимодействующим атомам. В 1917 В. Коссель выдвинул гипотезу, согласно которой электронная пара связи переходит целиком к одному из атомов с образованием ионной пары катион — анион, удерживающихся в молекуле электростатическими силами. Согласно обеим гипотезам наиболее устойчивыми оказываются соединения, в которых валентные электроны распределялись так, чтобы каждый атом был окружен оболочкой, имитирующей электронную оболочку ближайшего инертного газа (правило октета). Гипотеза Льюиса положила начало электронной теории ковалентной связи и ковалентности, гипотеза Косселя — теории ионной связи и гетеровалентности. Обе представляли крайние случаи общей картины полярной связи, когда электронная пара смещена к одному из атомов лишь частично и степень смещения может варьировать от 0 до 1 (см. Полярность химических связей ). В. атома в соединении, согласно классической электронной теории, равна числу его неспаренных электронов, участвующих в связях, а максимальная В. — обычно полному числу электронов в его валентной оболочке, то есть номеру труппы периодической системы, в которой находится элемент. Элементы одинаковых групп имеют одинаковое число валентных электронов, а внутри одинаковых подгрупп — и одинаковые или очень близкие электронные конфигурации (см. раздел 3). Сходство строения валентных оболочек атомов обусловливает сходство их соединений.
Ковалентность и гетеровалентность отражают специфику соответствующего типа химической связи. Для ковалентности важна насыщаемость связей, обусловливающая существование молекул в виде дискретных частиц с определённым составом и структурой. Ковалентность эффективна для органических и большинства простых неорганических соединений. Напротив, в случае гетеровалентности максимальное число ионов противоположного знака, способное разместиться вокруг данного иона, в основном определяется соотношениями их размеров. Ионная В. эффективна для сравнительно ограниченного класса соединений, в основном для различных солей щелочных, щёлочноземельных и некоторых др. металлов.
В. в комплексных соединениях. Ещё в конце прошлого века было найдено (А. Вернер , 1893), что многие соединения, как с максимальными (насыщенновалентные), так и с промежуточными В., типа ВСl 3, SiCl 4, PCl 5, CrCl 3и т.п., обладают склонностью к взаимодействию с другими насыщенновалентными соединениями — солями, окислами, молекулами типа H 2O, NH 3и др., с образованием довольно прочных комплексных соединений — K [BCl 4], K 2[SiCl 6], NH 4[PCl 6] и т.д. Исследования их строения рентгеновскими методами показали, что в комплексных анионах  и катионах
и катионах 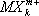 атомы лигандов Х обычно находятся в вершинах правильных многоугольников (октаэдра, тетраэдра и др.), а все связи М — Х одинаковы.
атомы лигандов Х обычно находятся в вершинах правильных многоугольников (октаэдра, тетраэдра и др.), а все связи М — Х одинаковы.
Для представлений о В. комплексные соединения необычны тем, что в них координационное число КЧ может быть больше общего числа валентных электронов атома М. Более того, в парамагнитных (см. Магнетохимия ) комплексах переходных и редкоземельных металлов — K 4[CrF 6], K 3[CrF 6], K 2[CrF 6] и др., некоторые электроны валентной оболочки остаются неспаренными и локализованными у центрального атома и практически не участвуют в связи. Классическая В. и КЧ, как правило, не совпадают, а способность к образованию октаэдрических и тетраэдрических. комплексов оказалась чрезвычайно распространённой и типичной для многих металлов и неметаллов, связанной сложной зависимостью с положением элемента в периодической системе и его В. в исходном простом соединении.
Поэтому было высказано предположение, что, наряду с «классической» В., которая реализуется в исходных простых соединениях типа ВСl 3, SiCl 4и др., атомы обладают также «координационной» В. (см. Донорно-акцепторная связь ), которая насыщается в комплексных соединениях (о природе координационной В. см. раздел 3). Предпринимались попытки описать связь в комплексных соединениях в рамках ионной теории, в которой считается, что анионы типа [PF 6] -и [МnO 4] -построены из ионов P 5++ 6F -и Mn 7++ 4O 2-и что В. центрального атома совпадает с зарядом его иона. Однако затраты энергии, необходимой для перевода 1 атома Mn и 4 атомов О в состояния Mn 7+и O 2-, далеко не компенсируются выигрышем в энергии при образовании связи. С появлением экспериментальных методов определения эффективных зарядов стало ясно, что эффективные заряды вообще редко превышают значения +1 или +2 у положительно заряженных и —1 у отрицательно заряженных атомов и обычно выражаются дробными долями заряда электрона (в перманганатном анионе заряд на Mn составляет лишь +1,5—+2,0 электрона). Поэтому ионная теория для большинства неорганических соединений, простых и комплексных, не может считаться корректной.
Успехи химии 20 в. и проблемы теории В. В 20 в. экспериментальной химией было синтезировано и изучено строение множества новых соединений, которые также оказалось невозможно уместить в рамки классических представлений о В. Оказалось, что склонность к образованию координационных соединений и насыщению координационных В. вообще чрезвычайно распространена и характерна практически для всех элементов и что суждения о В. на основании одного лишь стехиометрического состава очень часто оказываются несостоятельными без точных данных о структуре соединения и геометрическом расположении ближайшего окружения рассматриваемого атома. По мере развития структурных методов (см. Электронография молекул , Рентгенография молекул ) стало известно, что многие соединения с простым брутто-составом (AlCl 3, PdCl 2, MoO 3и др.), ранее считавшиеся простыми, в действительности даже в пара'х имеют димерное и полимерное строение — Al 2Cl 6, (PdCl 2) x( рис. 1 , а, б), (MoO 3) 2-5. В них «мостиковые» лиганды, соединённые одинаковыми связями с двумя атомами металлов (на рис. 1 они помечены цифрой 2), обладают координационным числом КЧ = 2. У соединений в твёрдом состоянии, которые часто построены ещё сложнее, КЧ галогенов и кислорода, ранее выбранного в качестве стандартного двухвалентного элемента, могут быть 3 и даже 4. В бороводородах каждый «мостиковый» атом водорода, считавшегося ранее стандартным одновалентным элементом, связан одинаковыми связями с двумя атомами бора ( рис. 1 , б). Алкильные группы также способны образовывать мостиковые связи в металлоорганических соединениях типа Al 2(CH 3) 6( рис. 1 , г) и др.
Читать дальшеИнтервал:
Закладка:









