Дмитрий Иванов - Нарушения обмена глюкозы у новорожденных детей

- Название:Нарушения обмена глюкозы у новорожденных детей
- Автор:
- Жанр:
- Издательство:неизвестно
- Год:неизвестен
- ISBN:нет данных
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Дмитрий Иванов - Нарушения обмена глюкозы у новорожденных детей краткое содержание
В работе представлен современный взгляд на нарушения обмена глюкозы у новорожденных детей. Описаны этиология, патогенез, клиника, основные методы лечения неонатальных гипо- и гипергликемий, неонатального сахарного диабета. Рассматривается влияние гипогликемии на нервно-психическое развитие.
Нарушения обмена глюкозы у новорожденных детей - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Таблица 19 Клинические особенности притранзиторном (ТСДН) и перманентном (ПСДН) сахарном диабете новорожденных (BrunkowM. Е. et al., 2001) [47]
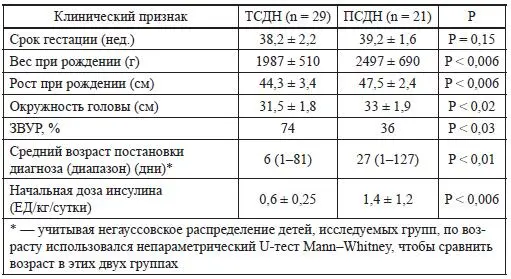
Хотя при обеих формах неонатального диабета достаточно часто встречаются пороки развития, но фенотипически дети отличаются (табл. 20).
Что касается, перманентного сахарного диабета, то в исследовании, проведенном IafuscoD. et al. (2002) [112] было продемонстрировано, что дети, заболевшие в первое полугодие жизни, в 6 раз чаще имели «защитные» аллели HLA-системы против классического диабета 1-го типа, по сравнению с детьми, заболевшими во втором полугодии. Кроме того, у детей, заболевших рано, в 5 раз реже встречались аутоиммунные маркеры.
Транзиторный сахарный диабет обычно развивается спорадически, хотя треть больных имеют установленную генетическую предрасположенность по линии отца. При этом не все отцы больны диабетом (von Muhlendahl К. Е., Herkenhoff Н., 1995) [155]. Некоторые больные имеют частичное удвоение длинного плеча 6-й отцовской хромосомы (6q24). В этой области находятся два гена (Arima Т. et al., 2001) [29]. Один кодирующий фактор транскрипции ZAC , регулирующий деление клетки и апоптоз, а также гипофизарную аденилатциклазу, активирующую полипептидный рецептор 1 и являющуюся мощным активатором секреции инсулина. Второй ген — HYMAI , функция которого в настоящее время неизвестна. В 2002 году было произведено экспериментальное моделирование ТСДН (Ма D. et al., 2002). В геном мыши был внедрен локус 6q24. У мышат развилось состояние, клинически очень сходное с транзиторным сахарным диабетом новорожденных, хотя и имевшее особенности.
Таблица 20
Фенотипические особенности детей при двух формах неонатального диабета
(Marquis Е. et al., 2002) [141]
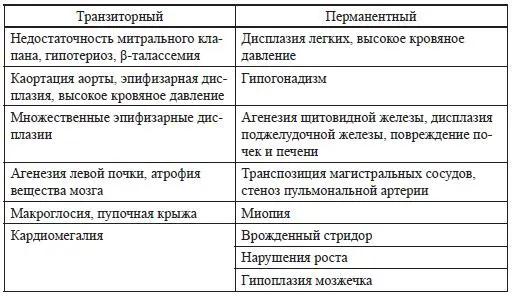
Отцовская передача данного локуса приводила к развитию гипергликемии и увеличению риска развития диабета у мышат. Интересно, что данный эксперимент установил уменьшение экспрессии ключевого фактора транскрипции PDX-1 в эмбриональной поджелудочной железе мышей. Однако конкретный клеточный дефект; приводящий к нарушению синтеза инсулина, остается неизвестным.
При перманентном сахарном диабете, по крайней мере, у 50 % больных установлена мутация в гене, кодирующем субъединицу АТФ-чувствительных калиевых каналов, находящихся в β-клетках поджелудочной железы. Наличие этой мутации приводит к тому что канал находится постоянно в открытом состоянии, что делает невозможным выделение инсулина (рис. 13–14). В настоящее время описано 9 мутаций в указанном гене. Некоторые из этих мутаций встречаются и при транзиторном сахарном диабете и диабете 2-го типа у более старших детей, что заставляет предполагать их генетическую общность (Gloyn A. L. et al., 2005 [86]; Babenko А. Р et al., 2006 [32]).
В настоящее время выделяют многочисленные наследственные синдромы, ассоциированные с перманентным сахарным диабетом новорожденных.
Агенезия поджелудочной железы и промоутер-фактор 1 инсулина (IPF1).Впервые ребенок с установленным нарушением гена, кодирующего промоутер-фактор 1 инсулина, и агенезией поджелудочной железы был описан Jonsson J. et al. в 1994 году [115]. Ребенок был гомозиготен по делеции единственного нуклеотида в 63 кодоне. Данный фактор регулирует эндокринное и экзокринное развитие поджелудочной железы. В 2000 году Vaulont S. et al. [203] показали, что он является регулятором инсулина и экспрессии гена соматостатина.
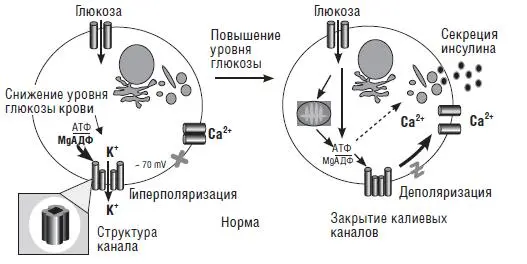
Рис. 13. Механизм секреции инсулина у здоровых людей(Polak М., Cave Н., 2007)
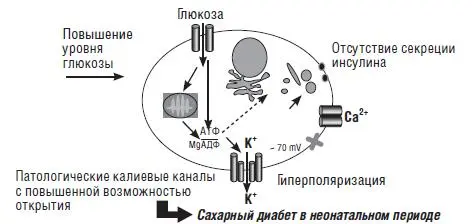
Рис. 14. Изменение секреции инсулина при генетически измененном калиевом канале(Polak М., Cave Н., 2007) [175]
Ранее Stoffers D. A. et al. (1998) [192] была описана семья, имевшая в 6 поколениях 8 больных с ранним началом диабета 2-го типа. Они были идентифицированы как гетерозиготы по той же мутации. Исследования, проведенные в последнее десятилетие во Франции, показали, что 6 % больных диабетом 2-го типа имеют мутации в гене, кодирующем промоутер-фактор 1 инсулина.
Гомозиготные аномалии в гене, кодирующем глюкокиназу.Мутации в указанном гене обычно приводят к умерено выраженной гипергликемии (Velho G., Froguel P., 1998) [204]. Глюкокиназа является регулятором метаболизма в β-клетках поджелудочной железы, контролирует уровень секреции инсулина. Njolstad P. R. et al. [161] в 2001 году описали две семьи (норвежскую и итальянскую), имевших больных различными типами диабета. В этих семьях родились дети, у которых в первый день жизни манифестировал классический перманентный сахарный диабет. При анализе оказалось, что они являются гомозиготами по мутации в гене, кодирующем глюкокиназу. Их родители были гетерозиготами по той же мутации и обладали пониженной толерантностью к глюкозе. Polak М., Cave Н. (2007) [175] считают, что если оба родителя имеют пониженную толерантность к глюкозе, то они должны быть обследованы на наличие мутаций в указанном гене.
Иммунная днзрегуляцня, полиэндокринопатия, энтеропатия, Х-связанная — IPEX-синдром.В 1996 году Peake J. Е. et al. [171] сообщили о мутации X хромосомы, сопровождающейся эксфолиативным дерматитом, диареей, гемолитической анемией, аутоиммунным поражением щитовидной железы и неонатальным диабетом. Дети умирают на первом году жизни от сепсиса. У некоторых детей описана агенезия островков Лангерганса. Аутоиммунная природа этого синдрома подтверждалась успешной терапией цитостатиками (Satake N. et al., 1993 [182]; Baud О. et al., 2001 [35]). Позже у больных были идентифицированы антитела к глутаминовой декарбоксилазе. Применение иммуносупрессивной терапии как этапа подготовки к трансплантации костного мозга приводило к купированию сахарного диабета, а затем диареи и дерматита. Установлено, что данная мутация, по крайней мере, в эксперименте, приводит к усиленной пролиферации СБ4+/СБ8-Т-лимфоцитов с мультиорганной инфильтрацией. В результате этого развивается гемофагоцитический синдром, и мальчики без лечения погибают на 15-25-й день жизни после рождения (Brunkow М. Е. et al., 2001) [47]. Описано три случая выздоровления после трансплантации красного костного мозга при данной патологии [35].
Синдром Wolcott-Rallison.Синдром назван в честь врачей Wolcott С. D., Rallison М. V. впервые в 1972 году, описавших трех больных в одной семье, имевших сочетание неонатального диабета и спондилоэпифизарных нарушений скелета [207]. Это аутосомно-рецессивное заболевание, характеризующееся ранним началом сахарного диабета, обычно в неонатальный период, и спондилоэпифизарными нарушениями роста. У больных часто отмечается гепатомегалия, задержка умственного развития, почечная недостаточность, ранняя смерть. В 2000 году Delepine М. et al. [66] описали двух подобных детей в разных семьях, имеющих мутацию 2р12. В данном локусе находится ген, являющийся регулятором синтеза инсулина и белков в β-клетках поджелудочной железы. В мире к 2010 году были описаны около 60 пациентов с синдромом Wolcott-Rallison (Julier С., Nicolino М., 2010) [116]. Хотя, конечно, больных небольшое количество, но, тем не менее, некоторые закономерности течения данного синдрома понятны. Сахарный диабет развивается в первые шесть месяцев жизни, спондилоэпифизарные нарушения роста начинают проявляться в первые два года жизни. Они выражены как клинически, так и рентгенологически (рис. 15–16).
Читать дальшеИнтервал:
Закладка:








