Чарльз Эллис - Эпигенетика

- Название:Эпигенетика
- Автор:
- Жанр:
- Издательство:Техносфера
- Год:2010
- Город:Москва
- ISBN:978-5-94836-257-1
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Чарльз Эллис - Эпигенетика краткое содержание
Книга ярко и наглядно повествует о новой науке общебиологического значения — эпигенетике, а также об ее отдельных областях. В издании представлено описание разных эпигенетических сигналов и механизмов их реализации, а также собственно феномен, история и концепции эпигенетики, ее отдельные механизмы и пути реализации эпигенетических сигналов в клетке. Авторы различных глав данной книги — ведущие в мире специалисты в области эпигенетики, являющиеся, как правило, и основоположниками ее отдельных областей.
Издание будет полезно широкому кругу читателей, интересующихся коренными проблемами живого мира, сущности жизни и молекулярных механизмов ее проявления.
По формирующейся традиции современной российской научной литературы, оригинальное русскоязычное печатное издание неопрятно переведено, отвратительно вычитано и содержит большое количество ошибок, начиная с обложки. Чарльз Дэвид Эллис указан как С. Д. Эллис.
Эпигенетика - читать онлайн бесплатно полную версию (весь текст целиком)
Интервал:
Закладка:
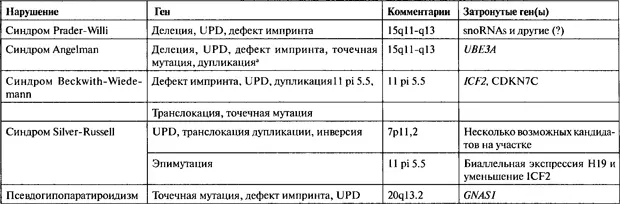
3. Заболевания человека
3.1. Нарушения геномного импринтинга
Открытие UPD было клинической «точкой входа» в нарушения геномного импринтинга у людей. В то время как PWS и синдром Ангелмана были первыми изученными нарушениями геномного импринтинга. синдром Беквита-Видемана (Beckwith-Wiedemann syndrome), псевдогипопаратироидизм и синдром Сильвера-Рассела (Silver-Russell syndrome) расширили этот список и поставили множество интригующих вопросов относительно того, как эпигенетические дефекты приводят к фенотипу, характерному для того или иного заболевания В следующем разделе мы даем краткий обзор клинических характеристик каждого нарушения, приводим разнообразные механизмы, ведущие к эпигенетипическим дефектам, а также описываем фенотипы и биологическую суть явлений, установленную в результате изучения этого типа нарушений (табл. 23.1).
Синдром Прадера-Уилли (PWS; OMIM 176270) и синдром Ангелмана (AS; OMIM 105830) в большинстве случаев вызываются одной и той же делецией 15q11-q13, размером 5—6 млн. нуклеотидов, но их фенотипы во многом различны. Геномный импринтинг в области 15q11-q13 объясняет эти фенотипические различия, с учетом того, что PWS вызывается делециями, унаследованными по отцовской линии, тогда как при AS делеции имеют материнское происхождение (Ledbetter et al., 1981; Magenis et al., 1987; Nicholls et al., 1989). PWS, который случается с частотой примерно 1/10000, был описан почти 50 лет назад и характеризуется младенческой гипотонией, задержкой развития, невозможностью роста вследствие плохого питания, апатией, за которыми следуют гиперфагия, тяжелое ожирение, низкоросл ость, вторичный гипогонадизм с гипоплазией гениталий и ослабление познавательных способностей. Пациенты с PWS также имеют отличительные физические черты, такие как небольшой размер кистей и стоп, миндалевидные глаза и тонкая верхняя губа. Большинство пациентов с PWS имеют слабое или среднее замедление умственного развития, подавляющее большинство из них проявляет разнообразные типы навязчиво-маниакального поведения, беспокойства, и иногда они замкнуты и несчастливы (рис. 23.4а). Наоборот, пациенты с AS имеют «счастливый настрой», часто улыбаются, и подвержены необъяснимым приступам смеха. Они страдают тяжелыми задержками развития, минимальными (если вообще имеют их) речевыми навыками. проблемами с равновесием (атаксией), производят аномальные похлопывающие движения кистями, характеризуются микроцефалией, припадками и некоторыми искаженными чертами, такими как выдающаяся нижняя челюсть и широкий рот (рис. 23.4б).
При обоих нарушениях могут наблюдаться гипотония, гипопигментация кожи и радужной оболочки и косоглазие. Большинство PWS и AS (-70%) вызваны отцовскими и материнскими делениями 15q11-q13, соответственно. Около 25% случаев PWS вызвано материнской UPD 15q11-q13, тогда как отцовская однородительская дисомия этого участка отвечает за 2—5% пациентов с AS (рис. 23.3). Разница в частоте однородительской дисомии между PWS и AS обычно инициируется материнским нерасхождением, поскольку на него влияет возраст матери, приводящий к зачатиям с трисомией или моносомией по хромосоме 15. Эти случаи затем подвергаются «спасению», что приводит к материнской UPD и PWS или отцовской UPD и AS, соответственно. Разница в частоте встречаемости этих двух однородительских дисомий предположительно связана с частотой образования двух аномальных яйцеклеток и с вероятностью «спасения» при обоих этих обстоятельствах. Транслокации в пределах критического PWS/AS участка отвечают менее чем за 10% таких случаев, но следует заметить, что такие транслокации связаны с высоким риском рецидива (до 50%), в зависимости от пола передающего родителя. Действительно, PWS и AS сосуществовали в некоторых семьях благодаря транслокациям или другим структурным аномалиям 15q11-q13, и фенотип при этом определялся полом передающего родителя (Hasegawa et al., 1984; Smeets et al., 1992).
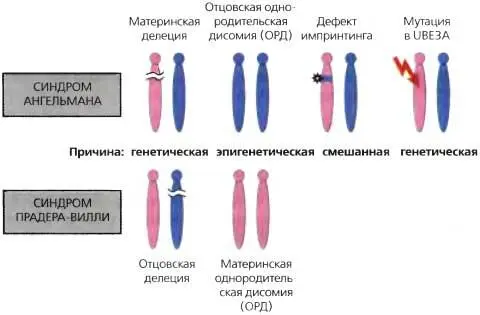
Рис. 23.3.Синдром Прадера-Вилли и синдром Ангелмана
Оба синдрома могут быть вызваны генетическими, эпигенетическими или смешанными дефектами
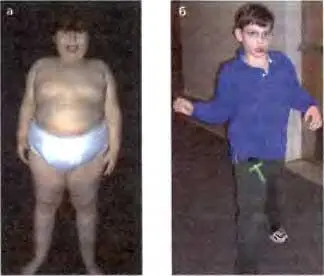
Рис. 23.4.Пациент с синдромом Прадера-Уилли (а) и пациент с синдромом Ангелмана (б)
Эти фотографии иллюстрируют существенные различия в клинической картине нарушений, вызванных дефектами в импринтированном участке. Фотографии любезно предоставлены Д-рами Daniel J Driscoll (а) и Carlos A. Bacino (б)
Дефекты импринтинга представляют еще один класс мутаций, ведущих к фенотипам PWS или AS. Такие дефекты, которые включают в себя разделенный на две части центр импринтинга (1 С) в пределах 15q11-q13 (Ohtaet al., 1999), являются причиной того, что хромосома, принадлежащая по происхождению одному из родителей, имеет измененный эпигенотип, как правило, эпигенотип хромосомы, происходящей от другого родителя. Дефекты импринтинга часто включают в себя делецию 1C, но есть случаи, когда такие дефекты оказываются имеющими место благодаря эпигенетической мутации, не затрагивающей последовательность нуклеотидов ДНК. Итог таких разнообразных нарушений импринтинга один и тот же, и он включает в себя изменения в метилировании ДНК, структуре хроматина и, в конечном счете, паттерны экспрессии генов. Дефекты импринтинга объясняют 2—5% случаев PWS и AS, а делеции в 1C обычно ассоциируются с 50%-ным риском рецидива, в зависимости от пола родителя, передающего аномалию, тогда как риск рецидива в семьях без делеции в 1C невысок. Идентификация дефектов импринтинга у небольшого количества пациентов с AS, которые были зачаты после инъекции спермы в цитоплазму яйцеклетки (ICSI, intracytolasmic sperm injection), поставила вопрос о том, что, возможно, этот способ оплодотворения in vitro вызывает дефекты импринтинга (Сох et al., 2002; Orstavik et al., 2003). Обнаружение дефектов импринтинга среди случаев AS у пациентов, родившихся у субфертильных родительских пар, не прошедших процедуру ICSI (но подвергнутых гормональной стимуляции), порождает дальнейшие вопросы относительно того, имеют ли бесплодие и дефекты импринтинга общие механизмы, или действительно вспомогательные репродуктивные технологии [гормоны и (или) ICSI] имеют эпигенетические последствия (Ludwig et al., 2005).
Какой конкретно ген (гены) затрагивается геномным импринтингом в 15q11-q13, известно лишь для AS, но не для PWS. Около 10—15% случаев AS вызываются мутациями потери функции в гене лигазы ЕЗ убиквитина (UBE3A ), кодирующем белок, связанный с Е6 (Е6-АР, E6-associated protein) (Kishino et al., 1997; Matsuura et al., 1997). Изучение экспрессии показало, что Ube3a экспрессируется исключительно с материнской аллели в мозжечковых клетках Пуркинье и в нейронах гиппокампа. Более того, мыши Ube3a + , лишенные материнской аллели, воспроизводят черты AS (Jiang et al., 1998). Эти результаты, так же как и данные по человеку, указывают на ген UBE3A, как на первопричину AS. Отцовская UPD или материнские делеции в 15q11-q13, ведут к потере экспрессии UBE3A в клетках Пуркинье. В случае дефектов импринтинга в 1C оказывается, что потеря сайленсинга антисмыслового транскрипта ведет к подавлению экспрессии гена UBE3A (Rougeulle et al., 1998). Интересно, что около 10% случаев AS остаются без молекулярного диагноза. Оказывается, что у ряда пациентов имеются мутации в белке ремоделинга хроматина — белке 2, связывающемся с метил-CpG, — что обсуждается ниже.
Читать дальшеИнтервал:
Закладка:







