Чарльз Эллис - Эпигенетика

- Название:Эпигенетика
- Автор:
- Жанр:
- Издательство:Техносфера
- Год:2010
- Город:Москва
- ISBN:978-5-94836-257-1
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Чарльз Эллис - Эпигенетика краткое содержание
Книга ярко и наглядно повествует о новой науке общебиологического значения — эпигенетике, а также об ее отдельных областях. В издании представлено описание разных эпигенетических сигналов и механизмов их реализации, а также собственно феномен, история и концепции эпигенетики, ее отдельные механизмы и пути реализации эпигенетических сигналов в клетке. Авторы различных глав данной книги — ведущие в мире специалисты в области эпигенетики, являющиеся, как правило, и основоположниками ее отдельных областей.
Издание будет полезно широкому кругу читателей, интересующихся коренными проблемами живого мира, сущности жизни и молекулярных механизмов ее проявления.
По формирующейся традиции современной российской научной литературы, оригинальное русскоязычное печатное издание неопрятно переведено, отвратительно вычитано и содержит большое количество ошибок, начиная с обложки. Чарльз Дэвид Эллис указан как С. Д. Эллис.
Эпигенетика - читать онлайн бесплатно полную версию (весь текст целиком)
Интервал:
Закладка:
Синдром Силвера-Рассела (SRS, Silver-Russell syndrome; OMIM 180860) — это нарушение развития, характеризующееся замедлением роста, невысокой фигурой, часто ассиметричной, и несколько бесформенными чертами лица и черепа, а также аномалиями пальцев. Наиболее заметная характеристика — это аномалии соматического роста, другие характеристики сильно варьируют. Генетически SRS гетерогенен, но подсчитано, что около 10% случаев являются результатом материнской UPD по хромосоме 7 (Eggermann et al., 1997). Предполагается, что SRS вызывается потерей функции гена, экспрессированного у отца (возможно, того, который способствует росту); но нельзя исключить альтернативную модель, а именно, сверхэкспрессию гена, подавляющего рост и экспрессированного у матери Интересно, что у некоторых индивидуумов с SRS была обнаружена эпигенетическая мутация, вызывающая деметилирование ICR1 на хромосоме 11р15. Этот эпигенетический дефект вызывает биаллельную экспрессию Н19 и пониженную экспрессию IGF2 (Gicquel et al., 2005).
Псевдогипопаратироидизм (РНР, pseudohypoparathyroidism) представляет собой группу фенотипов, являющихся результатом функционального гипопаратироидизма, несмотря на нормальный уровень паратироидного гормона (РТН, parathyroid hormone). Такие пациенты устойчивы к РТН (паратироидному гормону). Существует несколько клинических подтипов — la, lb, 1с, II и наследуемая остеодистрофия Олбрайта (OMIM 103580). Кроме функционального псевдогипопаратироидизма и остеодистрофии эти клинические варианты могут проявлять ряд соматических дефектов и дефектов развития. Гетерогенные с клинической точки зрения фенотипы являются результатом мутаций в гене GNAS1, кодирующем полипептид 1 (G sa) — белок, обладающий a-стимулирующей активностью и связывающийся с гуанином. GNAS1 картируется в хромосоме 20q 13.2. Локус GNAS1 имеет три альтернативных первых экзона, расположенных «вверх по течению» (экзоны 1А, XL, и NESP55), которые сплайсированы с экзонами 2-13 и производят разные транскрипты, и в случае с NESP55 и XL этот альтернативный сплайсинг продуцирует уникальные белки Около этих экзонов имеются дифференциально метилированные участки, принуждающие NESP55 экспрессироваться исключительно с материнских аллелей, тогда как XL, экзон 1А и антисмысловой транскрипт для NESP55 экспрессируются у отца. Хотя транскрипт, кодирующий белок G sa, экспрессирован биаллельно, в некоторых тканях, таких как проксимальные почечные канальцы, преимущественно экспрессирована материнская аллель. Сочетание геномных и тканеспецифичных импринтингов объясняет изменчивые фенотипы и эффект происхождения от одного или другого родителя даже для тех мутаций, которые имеют ясный аутосомный доминантный паттерн наследования (Hayward et al., 1998). Следует отметить, что у одного пациента с отцовской UPD участка GNAS1 развилось заболевание по типу lb (Bastepe et al., 2003).
Изучение генотипов и фенотипов этих клинических нарушений показало, что за исключением SRS все остальные геномные нарушения импринтинга (PWS, AS, BWS, и РНР) могут быть вызваны смесью генетических или эпигенетических аномалий, либо возникших de novo, либо унаследованных. Трудно поверить, что такая смешанная генетическая модель заболевания будет оставаться уникальной для этого небольшого набора нарушений. Немногим более десятилетия назад UPD была лишь теоретической возможностью, но сейчас установлено, что она имеет место на многих участках хромосом и приводит к разнообразным заболеваниям и фенотипам, касающимся развития. Одна из задач исследований генетики человека — выявить, какие гены отвечают за те или иные из ассоциированных с UPD фенотипов с целью установить список болезней, которые, вероятно, являются результатом смешанных генетикоэпигенетических механизмов.
3.2. Нарушения, влияющие на структуру хроматина в trans-конфигурации
Значение тонко отлаженной структуры хроматина для здоровья человека выдвинулось на первое место в связи с быстро растущим списком заболеваний человека, вызванных мутациями в генах, кодирующих белки, необходимые для структуры и ремоделинга хроматина. Сами по себе эти нарушения не являются эпигенетическими мутациями, но они изменяют состояния хроматина, являющиеся критическими компонентами эпигенотипа. Значительные различия между фенотипами, так же как тот факт, что едва уловимые изменения в уровне белка или даже консервативная аминокислотная замена могут приводить к заболеванию человека, все эти данные уже дают ключ к проблеме, касающейся строго контролируемой регуляции и взаимодействий белков ремоделинга хроматина. Нарушения, которые влияют на хроматин в trans -конфигурации, являются результатом либо нарушения функции белков, непосредственно участвующих в ремоделинге хроматина (таких, как белок, связывающийся с CREB, или СВР, ЕР300, а также белок, связывающийся с метил-CpG, или МеСР2), либо потери функции белков, участвующих в метилировании ДНК (таких, как de novo ДНК-метилтрансфераза ЗВ. DNMT3B, или метилентетрафолатредуктаза, MTHFR) (табл. 23.2). Нарушение функции любого из этих генов вызывает комплексные мультисистемные фенотипы или неоплазии, благодаря эффектам мисрегуляции экспрессии большого числа генов-мишеней, находящихся «ниже по течению». Вполне возможно, что существует множество заболеваний, которые вызываются мутациями в некодирующих РНК, действующими в trans -конфигурапии, хотя их еще предстоит обнаружить.
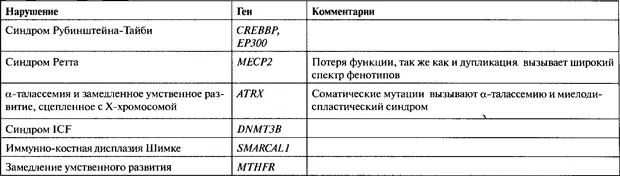
Синдром Рубинштейна-Тайби (RSTS, Rubinstein-Taybi syndrome; OMIM 180849) характеризуется задержкой умственного развития, широкими большими пальцами рук и ног, аномалиями лица, врожденными пороками сердца и повышенным риском образования опухолей. Высокая частота конкордантности у монозиготных близнецов, наряду с несколькими случаями передачи от матери к плоду, предполагают, что это заболевание имеет генетическую основу, и что наиболее вероятно аутосомное доминантное наследование. У некоторых пациентов с RSTS были установлены цитогенетические аномалии, включающие 16р13.3 (Tommerup et al., 1992), которые картируются в участке, содержащем ген белка, связывающегося с CREB (CREBBP, или СВР). Гетерозиготные мутации в CREBBP демонстрируют, что гапло-недостаточность СВР вызывает RSTS (Petrij et al., 1995). СВР впервые был описан как коактиватор реагирующего на с АМР белка CREB При повышении внутриклеточных уровней сАМР протеинкиназа А (РКА) перемещается в ядро и фосфорилирует CREB, что приводит к его активации и связыванию его с элементами ответа на количество сАМР (Мауг and Montminy, 2001). СВР — это крупный белок (~250 кДа) с бромодоменом, который, как показано, связывает РКА-фосфорилированный CREB (Chrivia et al., 1993). СВР, в свою очередь, активирует транскрипцию с промотора, содержащего CRE через ацетилирование всех четырех коровых гистонов в соседних нуклеосомах (Ogryzko et al, 1996). Кроме того, СВР взаимодействует непосредственно с основным транскрипционным фактором TFIIIB через участок на своем карбоксильном конце (Anas et al., 1994; Kwok et al., 1994). Функциональный анализ in vitro одной из миссенс-мутаций СВР (замена Arg-1378 на пролин), вызывающей RSTS, выявил, что эта мутация подавляет гистон-ацетилтрансферазную (HAT) активность СВР (Murata et al., 2001). Эти данные, в сочетании с тем, что мыши, гаплонедостаточные по СВР, имеют ослабленную память и обучаемость, измененную синаптическую пластичность и аномальное ацетилирование хроматина, поддерживают вывод о том, что ослабленная НАТ-активность СВР — это главная причина RSTS фенотипа (Alarcon et al., 2004). В соответствии с ролью, которую играет в заболевании понижение НАТ-активности, находится недавнее открытие того, что мутация во втором гене, рЗОО, кодирующем эффективную HAT и транскрипционный коактиватор, вызывает некоторые случаи RSTS (Roelfsema et al., 2005). Выявление того, что некоторые дефекты синаптической пластичности, как и недостатки обучаемости и памяти у СВР " мышей, могут быть ревертированы с помощью ингибиторов деацетилазы гистонов (HDAC) (Alarcon et al., 2004), вызывает вопрос о том, может ли лекарственная терапия, использующая эти реагенты, устранить некоторые ментальные проблемы при RSTS.
Читать дальшеИнтервал:
Закладка:







