Джеймс Уотсон - ДНК. История генетической революции

- Название:ДНК. История генетической революции
- Автор:
- Жанр:
- Издательство:Издательство Питер
- Год:2018
- Город:Санкт-Петербург
- ISBN:978-5-4461-0549-6
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Джеймс Уотсон - ДНК. История генетической революции краткое содержание
ДНК. История генетической революции - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Вполне возможно, что это и есть ключевая подсказка о сути патогенетического пути развивития шизофрении, но позволит ли она фармацевтам подступиться к созданию лекарства от этой болезни? Я отчаянно надеюсь, что так и будет. Все, о чем я сейчас говорю, всего лишь одно из проявлений фундаментальной проблемы, которая касается исследования всевозможных генетических заболеваний: как редких, так и распространенных. Как только мы найдем все или почти все аллели, обусловливающие эти болезни, возникнет другой вопрос: что делать дальше?
Иногда генетические болезни можно излечить без помощи новейших препаратов или специальной терапии, а просто поняв базовую причину такого заболевания. Возьмем, к примеру, один из наиболее изученных врожденных дефектов метаболизма, тот самый, из-за которого на некоторых продуктах, особенно на бутылках с газировкой, мелким шрифтом предупреждают: содержит фенилаланин. Фенилаланин – это аминокислота (один из основных первокирпичиков, слагающих белки), которая не усваивается людьми с генетическим расстройством под названием фенилкетонурия (ФКУ).
Эта история началась в Норвегии в 1934 году. Молодая мама была полна решимости выяснить, что же не так с двумя ее детьми четырех и семи лет, которые, казалось бы, родились совершенно здоровыми. Старший был плохо приучен к горшку и едва выговаривал несколько слов, а уж о полноценных предложениях не шло и речи. Врач и биохимик Асбьерн Феллингвыявил у этих детей любопытную биохимическую аномалию: у них в моче было слишком много фенилаланина. Асбьерн Феллинг обнаружил еще тридцать четыре таких случая в двадцати двух семьях по всей Норвегии и понял, что столкнулся с генетическим заболеванием.
На сегодняшний день уже известно, что ФКУ обусловлена мутацией в гене фенилаланингидроксилазы – это фермент, преобразующий фенилаланин в другую аминокислоту, тирозин. Это редкое наследственное заболевание, имеющее аутосомно-рецессивный путь передачи, в Северной Америке встречается примерно 1 раз на 10 тысяч человек. У детей, страдающих этой болезнью, фенилаланин накапливается в крови, тормозит развитие мозга и приводит к тяжелому отставанию в умственном развитии. Предотвратить заболевание просто: дети с ФКУ вырастают нормальными, если с самого рождения живут на диете, бедной фенилаланином, то есть с минимальным содержанием белка и без искусственно подслащенных напитков. Принципиально важно как можно раньше, лучше после рождения выяснить, рискует ли ребенок заболеть ФКУ. Роберт Гатри разработал простой анализ крови, позволяющий определить уровень фенилаланина, и неустанно продвигал его, пока анализ не вошел в стандартную практику медицинского скрининга. С 1966 года в США у каждого новорожденного берут анализ крови из пятки и проверяют уровень фенилаланина. Тест Гатри, при котором не проверяется ни единой «буквы» ДНК, ежегодно позволяет выявить у миллионов младенцев редкие генетические заболевания, в том числе ФКУ. До появления этого анализа 1 % всех случаев умственной отсталости в США связывали с ФКУ; теперь выявляется всего несколько случаев в год.
К сожалению, не по всем болезням и не во всех штатах проводится такой скрининг. В 2005 году Джим Келли, бывший квотербек в команде «Буффало Биллз» (американский футбол), и его жена Джил потеряли сына Хантера, который скончался от редкого генетического расстройства – болезни Краббе. Подобное заболевание показано в фильме « Масло Лоренцо », оно поддается лечению при условии ранней диагностики. Семья Келли основала фонд «Надежда Хантера», позволивший собрать миллионы долларов на исследование болезни Краббе и более полный скрининг новорожденных. Однако в масштабах такого огромного государства, как США, лишь в Нью-Йорке и еще нескольких штатах младенцев обязательно проверяют на болезнь Краббе, что является весьма постыдным фактом.
Через пятьдесят лет после появления теста забора крови из пятки у новорожденных Роберт Грин и его коллеги из Гарвардской медицинской школы запустили Baby Seq – рандомизированное клиническое исследование, в рамках которого планируется отсеквенировать геномы более сотни новорожденных и отследить в них 1700 заболеваний, начинающихся в детском возрасте. Для Гарварда это всего лишь небольшое клиническое исследование, но оно даст возможность совершить огромный скачок в оценке пользы от всеобщего скрининга новорожденных.
На 1950-е годы пришлось активное развитие цитогенетики – изучения хромосом под микроскопом. В диагностической практике этот подход показал, что при нарушении числа хромосом, когда их больше или меньше нормы, неизбежно возникают тяжелейшие болезни. Проблемы связаны с дисбалансом числа генов, то есть с отклонением от нормы «по два гена от каждого». Подобные расстройства не передаются по наследству, как миодистрофия Дюшенна или муковисцидоз, но все равно являются по сути генетическими; они возникают спонтанно при сбоях в клеточном делении и при образовании дефектных сперматозоидов и яйцеклеток.
Наиболее известная из таких болезней – синдром Дауна. Названа болезнь в честь Джона Лэнгдона Дауна, который в 1866 году впервые описал его характерные клинические признаки. «…Это [внешнее сходство] столь выраженное, что, если посадить таких детей бок о бок, сложно поверить, что они из разных семей». Девяносто лет спустя французский врач Жером Лежен обнаружил, что у детей с синдромом Дауна по три экземпляра одной хромосомы (впоследствии выяснилось, что лишняя 21-я хромосома). Среди генетиков-профессионалов такое расстройство именуется «трисомия 21».
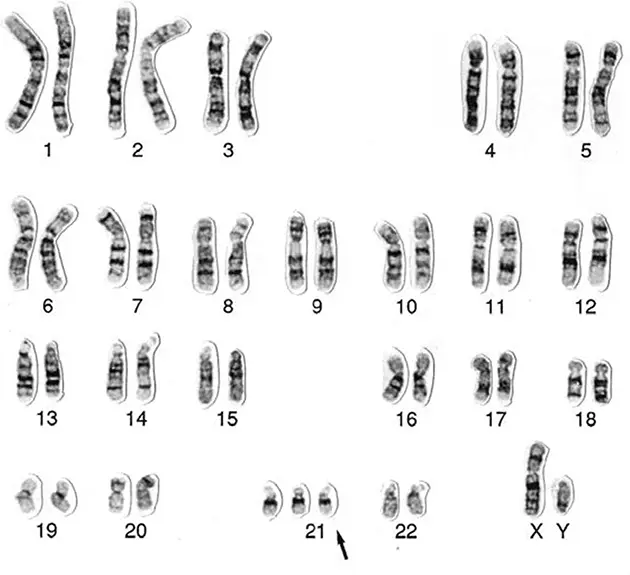
Трисомия 21 (синдром Дауна). Кариотип (полный набор хромосом), взятый у мужчины с синдромом Дауна. Обратите внимание на лишнюю 21-ю хромосому
Чем старше мать, тем выше риск синдрома Дауна. Когда матери 20, вероятность родить такого ребенка составляет примерно 1:1700, когда 35 – уже 1:400, а в 45 достигает 1:30. Именно поэтому многие старородящие женщины выбирают пренатальную диагностику, позволяющую определить, есть ли у плода тройная 21-я хромосома. Сегодня такой анализ входит в стандарт обследования беременных женщин старше 35 лет.
В Великобритании 30 % беременностей с синдромом Дауна обнаруживается при плановом тестировании у 5 % самых старородящих женщин. Этот метод выделяется своей высокой эффективностью по соотношению «число обнаруженных случаев на каждый потраченный фунт стерлингов», но что насчет остальных 70 % случаев синдрома Дауна? Неинвазивные методы являются альтернативными умеренно рискованным технологиям: амниоцентезу и биопсии хориона, – меняют профиль пренатальной диагностики. В конце 1990-х годов различные исследователи, в частности Деннис Ло из Китайского университета в Гонконге, показали, что ДНК плода можно обнаружить в плазме крови матери. Десять лет спустя Ло и группа ученых из Стэнфордского университета под руководством Стивена Квейка независимо показали, что анализ такой ДНК позволяет выявить трисомию 21. Процедура под названием «неинвазивное пренатальное тестирование» (NIPT) относительно проста: исследователь секвенирует 5–10 миллионов коротких произвольно взятых фрагментов ДНК из материнской плазмы, а затем сличает их с соответствующими хромосомами. Если в ДНК плода обнаруживается лишняя 21-я хромосома, то из 21-й хромосомы будет больше фрагментов, чем из других. Аналогично обнаруживаются трисомии и в других хромосомах, в частности в 13-й или 18-й. Такие трисомии вызывают соответственно синдром Эдвардса и синдром Патау, тяжелые генетические расстройства; ребенок с такими болезнями обычно умирает спустя несколько недель или месяцев после рождения. Другие трисомии летальны на пренатальном этапе, так прерывается около 30 % беременностей, и около половины подобных случаев прерывания беременности связаны с теми или иными хромосомными аберрациями.
Читать дальшеИнтервал:
Закладка:









