Андрей Обрезан - Семейная медицина

- Название:Семейная медицина
- Автор:
- Жанр:
- Издательство:Литагент СпецЛит
- Год:2008
- ISBN:978-5-299-00406-9
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Андрей Обрезан - Семейная медицина краткое содержание
Книга раскрывает специфику семиотики, диагностики основных состояний, встречающихся в практике врача. Изложенные сведения призваны способствовать усвоению навыков дифференциальной диагностики заболеваний внутренних органов, получению знаний о современном арсенале диагностических методов, помогут специалисту оценить симптомы и синдромы, правильно сформулировать диагноз, определить перечень дифференцируемых заболеваний, спланировать характер проводимой терапии.
Содержание и объем пособия соответствуют учебной программе для медицинских вузов и факультетов подготовки врачей по разделу «Семейная медицина».
Семейная медицина - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Синдром Ашера ( Ascher, синдром Laffer – Ascher ) – сочетание блефарохалазиса со струмой и с так называемой двойной губой (аутосомно-доминантное наследование): блефарохалазис; двойная губа в связи с образованием складок слизистой оболочки; постоянное или рецидивирующее припухание губ в результате хронического отека; струма, обычно без эндокринных нарушений.
Синдром Бадда – Киари (см. подразд. 8.3).
Синдром Иценко – Кушинга (см. подразд. 6.3).
Синдром Клайнфельтера ( Klinefelter , синдром Klinefelter – Reifenstein – Albright ) – наследственное выпадение канальцевой функции яичек в период полового созревания (сцепленное с Х-хромосомой наследование, генотип ХХY): гинекомастия (не всегда) у нормально развитого в остальном юноши; гипоплазия яичек, азоо– и олигоспермия; увеличенное количество гонадотропина и 17-кетостероидов в моче; нормальное вторичное оволосение. Изредка – евнухоидный облик. Нередко снижен основной обмен. Хромосомный пол женский. Болезнь проявляется в период полового созревания.
Синдром Лафта ( Luft ) – симптомокомплекс у больных с наследственными аномалиями митохондрий поперечнополосатой мускулатуры (возможно аутосомно-рецессивное наследование): профузный пот, полидипсия без полиурии, полифагия, прогрессирующее исхудание, астения, мышечная слабость и атрофия, патологическая электромиограмма, отсутствие глубоких сухожильных рефлексов, креатинурия. Значительно повышен основной обмен.
Синдром Лоуренса – Муна – Барде – Бидля – наследственная болезнь, характеризующаяся ожирением, гипогенитализмом, пигментным ретинитом, задержкой умственного развития, полидактилией и синдактилией; предположительно наследуется по аутосомно-рецессивному типу.
Синдром мальабсорбции – сочетание гиповитаминоза, анемии и гипопротеинемии, обусловленное нарушением всасывания в тонкой кишке.
Синдром Марфана (см. подразд. 6.3).
Синдром Пейджа ( Page, diencephalosis ) – диэнцефальная форма ювенильной гипертонии: нестабильная ювенильная гипертензия; периодически на груди появляются красные пятна с неровными краями; нередко в этой области наблюдается гипергидроз. Часто – увеличенная щитовидная железа; несколько повышен основной обмен. Струмэктомия не дает эффекта. Выраженный гинекотропизм.
Синдром Прадера – Вилли ( Prader – Willi , синдром Prader – Labhart – Willi , HHHO ( hypotonia, hypomentia, hypogonadismus, obesitas ), синдром Royer )– комплекс наследственных аномалий (возможно, аутосомно-рецессивное наследование): акромикрия, низкий рост, ожирение; мышечная гипотония (постепенно исчезающая); гипогенитализм. Симптомы проявляются вскоре после рождения. Позднее присоединяются признаки олигофрении и сахарного диабета (с доброкачественным течением). Реже наблюдается также синдактилия, высокое нёбо.
Синдром Труэлля – Жюне ( Troell – Gunet , гипертиреоз акромегалоидный с гиперостозом) – сочетание диффузного гиперостоза свода черепа, акромегалии и признаков гиперфункции щитовидной железы, обусловленное увеличенной секрецией аденогипофизом соматотропного и тиреотропного гормонов.
Синдром Фелти ( Felty, neutropenia splenica et arthritis rheumatoides, splenomegalia-leukopenia-arthritis ) – форма ревматоидного артрита: ревматоидный полиартрит, увеличенная селезенка, лейкопения, гранулоцитопения, тромбоцитопения, анемия, гипер– и диспротеинемия. В более поздних стадиях – генерализованное припухание лимфатических узлов; открытие части тела с желто-коричневой пигментацией; ахилия, изъязвления в слизистой оболочке полости рта. В костном мозге – пангемоцитопения, ретикулоцитоз. Выраженный гинекотропизм.
Синдром Шегрена (см. подразд. 11.3). Синдром Шмидта (см. подразд. 6.3).
Субсепсис Висслера – Фанкони ( Wissler – Fanconi, синдром Wissler, синдром Fanconi – Wissler, subsepsis allergica, sepsis hyperergica, pseudosepsis allergica ) – хроническое, септически-гиперэргическое общее заболевание с кожными аллергическими проявлениями: интермиттирующая или постоянная лихорадка, длящаяся месяцами; постоянно рецидивирующая кратковременная экзантема; иногда развиваются подкожные узелки и появляется ревматоидное припухание мягких тканей; часто мио– или перикардит; иногда увеличиваются лимфатические узлы; селезенка увеличивается редко; гемокультура отрицательна. В крови нейтрофилез со сдвигом влево, изредка эозинофилия. Болеют преимущественно маленькие дети.
ГЛАВА 15
ПОЛИСИСТЕМНЫЕ И ПОЛИМЕТАБОЛИЧЕСКИЕ ЗАБОЛЕВАНИЯ
15.1. Классификации некоторых полисистемных и полиметаболических заболеваний
Антифосфолипидный синдром (АФС) представляет собой клинико-лабораторный синдром, проявляющийся рецидивирующими тромбозами (артериальными и/или венозными), привычным невынашиванием беременности, тромбоцитопенией и циркуляцией в крови антифосфолипидных антител.
1. АФС у больных с достоверным диагнозом СКВ (вторичный АФС).
2. АФС у больных с волчаночноподобными проявлениями.
3. Первичный АФС.
4. «Катастрофический» АФС (острая диссеминированная коагулопатия/васкулопатия) с острым мультиорганным тромбозом.
5. Другие микроангиопатические синдромы (тромботическая тромбоцитопеническая пурпура/гемолитико-уремический синдром); HELLP-синдром (гемолиз, повышение активности печеночных ферментов, снижение содержания тромбоцитов, беременность); ДВС-синдром; гипопротромбинемический синдром.
6. «Серонегативный» АФС.
Рассмотрим диагностические критерии АФС (табл. 97).
Таблица 97
Диагностические критерии антифосфолипидного синдрома
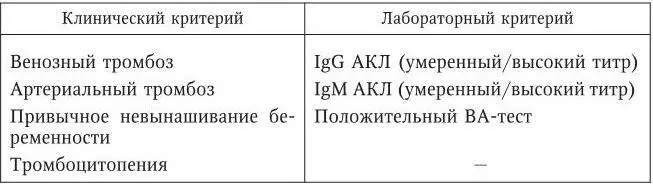
Для диагноза АФС необходимо наличие по крайней мере одного (любого) клинического и одного (любого) лабораторного признака; АФЛА должны выявляться не менее двух раз в течение 3 мес.
Лабораторная диагностика АФС основана в первую очередь на определении ВА с помощью функциональных тестов и АКЛ иммуноферментным методом с использованием иммобилизованного на твердой фазе кардиолипина. АКЛ могут относиться к различным изотипам иммуноглобулинов (IgG, IgM, IgA). Частота обнаружения IgG АКЛ в сыворотках здоровых людей варьируется от 0 % до 14 %. Таким образом, однократное обнаружение АФЛ не может служить основанием для постановки диагноза АФС.
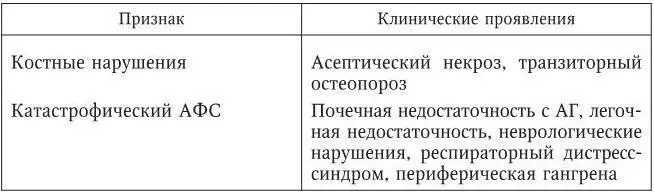
Основные клинические проявления АФС следующие (табл. 98).
Таблица 98
Основные клинические проявления антифосфолипидного синдрома

Метаболический синдром – совокупность клинических, лабораторных и инструментальных признаков, объединенных единым патогенезом, ускоряющим коморбидное течение ишемической болезни сердца, артериальной гипертонии, абдоминального ожирения и нарушений углеводного обмена.
Читать дальшеИнтервал:
Закладка:









