Коллектив авторов - Эндокринология. Большая медицинская энциклопедия

- Название:Эндокринология. Большая медицинская энциклопедия
- Автор:
- Жанр:
- Издательство:Array Литагент «5 редакция»
- Год:2014
- Город:Москва
- ISBN:978-5-699-45353-5, 978-5-699-45342-9
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Коллектив авторов - Эндокринология. Большая медицинская энциклопедия краткое содержание
Отеки под глазами, снижение памяти, резкое похудание или прибавка в весе, нарушение аппетита, жажда, нарушение менструальной функции, синдром компьютерной усталости – все это тоже может быть симптомом гормонального нарушения.
В данной книге читатель найдет всю информацию об эндокринной системе человека и подробное описание болезней, связанных с ее изменениями.
Энциклопедия предназначена для практикующих врачей и студентов медицинских вузов.
Эндокринология. Большая медицинская энциклопедия - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Чтобы подтвердить недостаточность секреции СТГ, используется не менее двух стимулирующих тестов.
Таблица №1
Дифференциально-диагностические данные при различных видах низкорослости
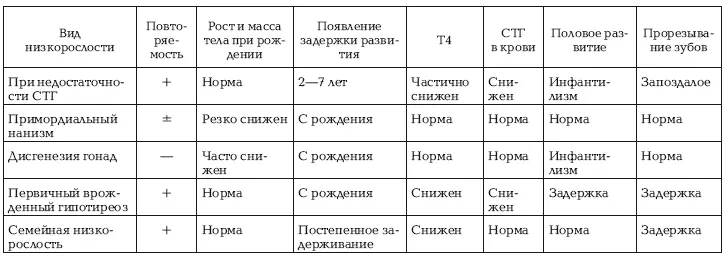
Гипофизарный нанизм дифференцируется от типичных генетических симптомов.
Прогерия (синдром Хатчинсона – Гилфорда) проявляется у детей к концу первого года жизни, когда выявляются задержка роста и прогрессирующая алопеция. Для детей с преждевременным старением характерны следующие симптомы:
1) большая голова с недоразвитой нижней челюстью;
2) маскообразное лицо с выраженным экзофтальмом, клювовидным носом;
3) узкая грудная клетка;
4) тонкая конечность с атрофией мышц;
5) ограничение подвижности в суставах;
6) отсутствие потовых и сальных желез;
7) позднее прорезывание зубов с аномальным разряжением;
8) отставание нервно-психического развития;
9) низкий уровень ИФР-1 при нормальной суточной секреции гормона роста;
10) увеличение суточной экскреции талу-роновой кислоты у детей до 10 – 20%.
Птицеголовые карлики (синдром Секкеля) имеют специфические черты, так как у них отмечается задержка роста во внутриутробном периоде. Специфические черты выражаются микроцефалией, низким расположением ушей, изменением пятого пальца, а также умственным недоразвитием.
При синдроме Прадера – Вилли появляется задержка роста с рождения, умственное недоразвитие, нарастание массы тела за счет ожирения, нарушение толерантности к углеводам. Данное заболевание характеризуется утратой парацентралярного района хромосомы 15.
При синдроме Лоуренса – Муна – Барде – Бидля определяются: низкий рост, гипогонадизм, дегенерация сетчатки пигментного характера, отставание в умственном развитии.
Ахондроплазия характеризуется:
1) диспропорциональным укорочением конечностей (плеч, бедер);
2) утолщением и укорочением пальцев;
3) поясничным лордозом;
4) круглой головой;
5) седловидным носом;
6) широкой переносицей;
7) рентгенологически выявляется дистрофия метафизов с бокаловидными участками разряжения костной ткани.
Наследуется по аутосомно-рецессивному типу.
Лечение
В раннем детском возрасте назначается заместительная терапия гормонами роста, она приближена к физиологическим условиям секреции соматотропина.
Для лечения используется гормон роста, полученный методом генной инженерии. Стандартная доза составляет 0,05 мг/кг массы тела ежедневно в 20 – 22 ч. Введение гормона роста увеличивают после первого года лечения, в последующие годы скорость роста снижается до 6 – 7 см в год.
Другая схема предусматривает введение гормона роста из расчета на квадратный метр площади тела, которая определяется по соответствующим формулам.
Введение препаратов в период полового развития проводится с увеличением дозировок препаратов в 2 раза.
Ряд препаратов вводится с помощью шприцев-ручек (нордитропин, генотропин, хуматрон и др.).
При недостаточной секреции гормона роста применяются препараты, которые стимулируют его секрецию (гексаредин).
Его назначение эффективно при ожирении, гипопитуитаризме. Введение в лечение глюкокортикоидов проводится при недостаточности глюкокортикотропного гормона.
Заместительное лечение осуществляется гонадотропином. При достижении костного возраста 11 – 12 лет девочкам назначают этинилэстрадиол из расчета 0,1 мкг/кг, мальчикам рекомендуется тестостерон внутримышечно из расчета 50 – 100 мг/м 2поверхности тела в месяц.
При недоразвитии половых органов у мальчиков назначается однократно внутримышечно тестостерон эпантант. Оценка результата проводится через месяц.
Эстрогены назначаются девочкам старше 16 лет: микрофоллин или этинилэстрадиол в первые 16 – 20 дней цикла, во вторую фазу цикла назначается прогестерон.
Мальчикам и мужчинам после закрытия зон роста назначаются гормоны (тестостерон пролонгированного действия, а также сусанон, тестенат по рекомендуемым схемам).
Женщинам назначаются постоянно половые гормоны (микрофоллин, эстрадиол) для появления вторичных половых признаков. У женщин такое лечение рекомендуется проводить до климакса.
Карликовый нанизм требует проведения общеукрепляющего лечения, с обязательным курсовым назначением препаратов кальция, фосфора, витаминов А и D. Лечение проводится с заменой менее эффективных препаратов на более эффективные.
При вторично возникшей карликовости, кроме лечения гормонами роста, требуется лечение основного заболевания.
Прогноз
Длительное лечение больных, страдающих недостаточностью гормона роста, часто дает положительные результаты, если начато своевременно лечение в детском возрасте.
Глютаминовая кислота, аминалон, церебролизин назначаются при нарушениях функции центральной нервной системы.
При гипотиреозе назначается тиреоидин, тироксин и другие препараты.
Больные с гипофизарным нанизмом находятся на диспансерном учете у врача-эндокринолога пожизненно.
В период медикаментозного лечения больные проходят обследования каждые 2 – 3 месяца. В остальных случаях каждые 6 – 12 месяцев производится контроль содержания соматотропного гормона в крови. По показаниям больные осматриваются узкими специалистами.
Глава 2.
Болезнь Иценко – Кушинга
Болезнь Иценко – Кушинга – заболевание, характеризующееся повышением продукции кортикостероидов вследствие увеличения выработки надпочечниками адренокортикотропного гормона. Это заболевание, связанное с поражением межуточно-гипофизарной области, описано в 1926 г. Н. И. Иценко, а как гипофизарный синдром его описал Г. В. Кушинг.
Болезнь Иценко – Кушинга связана с избыточной секрецией кортикотропина при поражении гипоталамо-гипофизарной системы, в результате чего развивается гиперплазия надпочечников.
Механизм и причины развития заболевания
Этиология болезни Иценко – Кушинга не определена. Различается истинная болезнь Иценко – Кушинга и синдром, носящий тоже название (гиперкортицизм). Развитие последнего возможно при злокачественной и доброкачественной кортикостероме. Он может возникнуть при гиперплазии коры надпочечников.
При болезни Иценко – Кушинга часто определяются опухоли гипофиза, которые возникают в различные возрастные периоды и при травмах головного мозга, после перенесенных нейроинфекций. В этих случаях возникает избыточная продукция кортиколиберина, образование которого стимулируется определенными нейронами. При длительном повышении продукций кортиколиберина возникает гиперплазия, переходящая в микроаденомы, аденому гипофиза.
Читать дальшеИнтервал:
Закладка:





![Коллектив авторов - Арахна [Большая книга рассказов о пауках]](/books/1059707/kollektiv-avtorov-arahna-bolshaya-kniga-rasskazov.webp)



