Андрей Брюховецкий - Боковой амиотрофический склероз

- Название:Боковой амиотрофический склероз
- Автор:
- Жанр:
- Издательство:неизвестно
- Год:неизвестен
- ISBN:9785005300577
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Андрей Брюховецкий - Боковой амиотрофический склероз краткое содержание
Боковой амиотрофический склероз - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Курение как патогенетический фактор БАС . Методы доказательной медицины подтвердили связь курения с развитием БАС (Chen Lien al., 2015).
На сегодня не существует общепризнанной гипотезы патогенеза бокового амиотрофического склероза. Согласно современным представлениям, развитие БАС обусловлено взаимодействием наследственных и экзогенных провоцирующих факторов. Множество патологических изменений в нейронах приводит к предположению о многовариантном этиологическом факторе.
Окислительный стресс считается важнейшим фактором в этиопатогенезе БАС. Известно, что окислительный стресс причастен к процессу нейродегенерации при БАС. Также известно, что скопление образцов активных форм кислорода вызывает гибель клеток. Поскольку мутации, происходящие в гене SOD1, могут привести к семейному типу БАС, этот механизм окислительного стресса дегенерации двигательных нейронов при БАС представляет научный интерес. Данная гипотеза поддерживается биохимическими изменениями, отражающими повреждения свободных радикалов и патологический метаболизм свободных радикалов в цереброспинальной жидкости и образцах аутопсий пациентов с БАС (Ferrante et al., 1997; Smith et al., 1998; Tohogi et al., 1999). Кроме того, у образцов фибробластов, культивированных у больных с БАС, выявлялась повышенная чувствительность к окислительным разрушениям по сравнению с участниками контрольных групп (Aguirre et al., 1998).
Предполагается, что перекись водорода может служить аномальным субстратом для конформированной молекулы SOD1. В результате происходит усиление пероксидантных реакций и возрастает производство токсичных гидроксильных радикалов. Существенная роль окислительного стресса в патогенезе БАС подтверждается биохимическими исследованиями, при которых у больных обнаружилась недостаточность ряда систем антиоксидантной защиты, дисфункции митохондрий, дисметаболизм глутатиона, эксайтотоксина глутамата и механизмы глутаматного транспорта. Возможно, окислительное повреждение белковых мишеней (SOD1, нейрофиламентных белков, альфа-синукленина и т.д.) может облегчать и ускорять их совместную агрегацию, формирование цитоплазматических включений, которые служат субстратом для дальнейших патохимических окислительных реакций.
Апоптоз – опосредованная каспазами гибель клеток . Предполагается, что окончательный процесс гибели двигательных нейронов при БАС напоминает апоптозный путь запрограммированной клеточной гибели. Апоптоз представляет собой механизм, посредством которого двигательные нейроны отмирают в моделях мутантных мышей с трансгенным SOD1. В этих моделях были последовательно активированы апоптозные сигналы каспазы-1 и -3. Хроническая активация каспазы-1 происходила в раннем пресимптоматическом периоде, а каспаза-3 была активирована намного позже, будучи финальным эффектором гибели клеток (Pasinelli, 2000). В соответствии с данными сообщениями, внутрицеребровентрикулярная доставка широкого ингибитора каспазы приводила к снижению mRNA уровней каспазы-1 и -3 в тканях спинного мозга мутантных мышей с трансгенным SOD1 (G93A). Кроме того, двигательные нейроны были сохранены у мышей, прошедших трансфузию ингибитором каспазы, в сравнении с мышами, получившими трансфузию носителя-растворителя. Также удалось задержать начало развития заболевания и его прогрессирование (Li et al., 2000). Биохимические маркеры апоптоза определяются в терминальной стадии БАС пациентов и животных моделей. Ключевые элементы пути здорового апоптоза вовлечены в гибель клеток при БАС, в том числе семейство каспаз протеолитических ферментов, Bcl2 семейство онкопротеинов (антиапоптозные и проапоптозные онкогены) и ингибирующее апоптоз семейство протеинов. Что касается дополнительного подтверждения, говорящего в пользу апоптозного механизма гибели нейронов, было замечено, что избыточная экспрессия антиапоптозного протеина Bcl-2 (Kostic et al., 1997) и удаление проапоптозного протеина Bax (Gould et al., 2006) способны удерживать и сохранять двигательную функцию и продлевать выживаемость мутантных мышей с трансгенным SOD1 (G93A). Таким образом, токсичность мутантного SOD1, по всей видимости, опосредуется, по крайней мере частично, каспазами и другими апоптозными факторами.
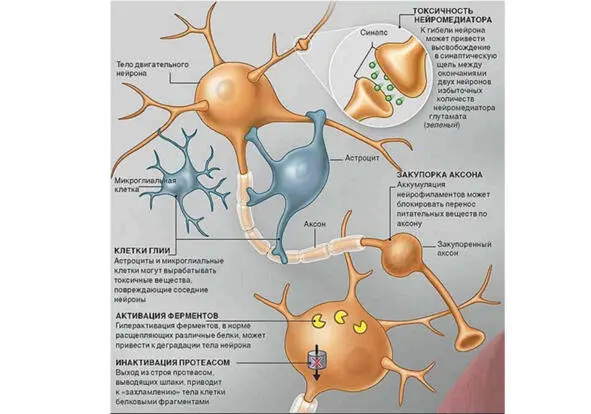
Рис. 1. Установленные негенетические молекулярно-биологические механизмы развития бокового амиотрофического склероза
В интернете мы нашли очень познавательную схему патофизиологических нарушений и молекулярно-биологических механизмов, происходящих в нервной ткани при БАС, и приводим ее для иллюстрации всего вышесказанного (рис. 1).
Представленная схема позволяет очень наглядно увидеть все основные молекулярно-биологические механизмы патогенеза данного заболевания (рис. 2).
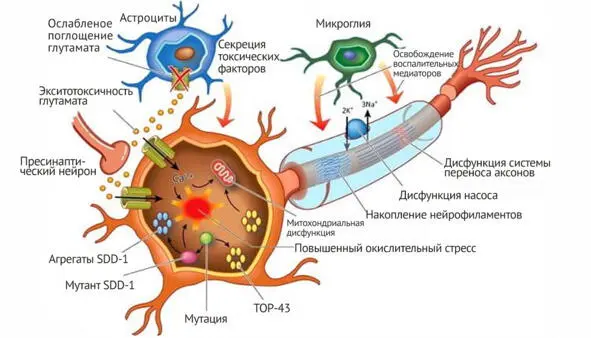
Рис. 2. Схема локальных молекулярных повреждений в двигательном нейроне при боковом амиотрофическом склерозе
Как видно на рисунке 2, локальные молекулярные повреждения в мотонейроне связаны с ослаблением поступления глютамата из астроцитов с формированием экситоксичности глютамата, что приводит к митохондриальной дисфункции в мотонейроне, нарушению обмена внутриклеточного Са 2+и возникновению повышенного окислительного стресса, появлению мутантного фермента СОД-1 и, как следствие, появлению мутаций в других генах ядра нейрона. Эти молекулярно-биологические нарушения приводят к появлению в цитоплазме мотонейрона агрегатов СОД-1, которые усиливают процессы мутагенеза. Освобождение воспалительных медиаторов из клеток микроглии приводит к локальному повреждению структуры аксонов и нарушает их информационную проводимость путем нарушения их цитоскелета.
Все большее число исследований указывают на то, что воспаление в центральной нервной системе, а также повышенная концентрация иммунных клеток, вызывающих воспалительные реакции, обнаруженная в центральной нервной системе пациентов с БАС, являются ключевыми этиопатогенетическими факторами при БАС. Клинические исследования выявили периферическое воспаление в БАС; были обнаружены такие маркеры воспаления, как Т-клетки, цитокины и хемокины. Китайские ученые из Университета Минзу в Китае (Minzu University of China; Пекин) провели систематический обзор и метаанализ 25 исследований, включая исследование 812 пациентов с БАС и 639 человек в контрольной группе. Это было сделано для устранения противоречивых результатов. Сравнивались уровни воспалительных цитокинов пациентов с БАС и пациентов контрольной группы, затем данные клинических исследований объединялись и анализировались. Метаанализ выявил значительную гетерогенность для 8 из 14 цитокинов. Интерлейкин (IL) -2, IL-4, IL-17, фактор роста эндотелия сосудов (VEGF) показали умеренные уровни гетерогенности, тогда как фактор некроза опухоли-α (TNF-α), моноцитарный хемоаттрактантный белок-1 (MCP-1), интерферон гамма (IFNγ), IL-5 показали высокие уровни гетерогенности. Анализ в подгруппах показал, что уровни TNF-α в крови были значительно увеличены у пациентов с БАС по сравнению с пациентами в контрольной группе. Ученые отметили: предыдущие данные показали, что уровни TNF-α, IL-6 и IL-1β в крови повышены у пациентов с болезнью Альцгеймера и болезнью Паркинсона, в то время как рецептор фактора некроза опухоли 1 (TNFR1) увеличен при болезни Паркинсона, что дает представление об общем механизме у различных нейродегенеративных нарушений. Авторы пришли к выводу, что метаанализ впервые использовался для исследования изменений уровней воспалительных цитокинов у пациентов с БАС и показал увеличение уровней TNF-α, TNFR1, IL-1β, IL-6, IL-8 и VEGF в периферической крови у пациентов с БАС по сравнению с пациентами в контрольной группе. В исследовании подчеркивается, что периферические уровни цитокинов могут быть биомаркерами для БАС, что может быть интересным для медицинских работников, стремящихся преодолеть разрыв между постановкой диагноза и появлением симптомов. Исследование было опубликовано 22 августа 2017 г. в журнале Scientific Reports (https://studfiles.net/preview/1818013/page:92/).
Читать дальшеИнтервал:
Закладка:








