Валерий Крылов - Каталитический риформинг бензинов. Теория и практика

- Название:Каталитический риформинг бензинов. Теория и практика
- Автор:
- Жанр:
- Издательство:неизвестно
- Год:2021
- ISBN:нет данных
- Рейтинг:
- Избранное:Добавить в избранное
-
Отзывы:
-
Ваша оценка:
Валерий Крылов - Каталитический риформинг бензинов. Теория и практика краткое содержание
В книге представлен анализ теоретических и практических положений технологии процесса каталитического риформинга бензиновых фракций. Изложен опыт и приведены лучшие практики эксплуатации современных установок риформинга.
Книга предназначена для инженерно-технических работников нефтеперерабатывающих заводов, проектных организаций, преподавателей и студентов вузов.
Каталитический риформинг бензинов. Теория и практика - читать онлайн бесплатно ознакомительный отрывок
Интервал:
Закладка:
Почему происходит увеличение скорости реакции циклизации при переходе от н-гексана к н-гептану.
Кинетический и термодинамический контроль реакций риформинга.
Структурно-чувствительные и структурно-нечувствительные реакции
Термодинамика устанавливает принципиальные ограничения на направление и максимальную глубину химического превращения. Равновесная степень химического превращения является предельно возможной величиной, которая может быть получена при условии достижения химического равновесия при данных давлении и температуре.
Реально достижимая степень превращения может быть ниже, если достижение химического равновесия невозможно из-за существования кинетических барьеров в виде высоких энергий активации реакции.
Зависимость константы скорости реакции от температуры описывается уравнением Аррениуса, где экспоненциальный множитель представляет собой долю молекул, обладающих кинетической энергией не менее Е апри данной температуре Т :
где K о – предэкспоненциальный множитель; Е а– энергия активации; R – универсальная газовая постоянная; Т – температура, К.
Значения энергии активации для ряда реакций платформинга, кДж/моль, представлены ниже:
– изомеризация парафиновых и нафтеновых углеводородов – 105,
– дегидрирование парафиновых и нафтеновых углеводородов – 84,
– дегидроциклизация парафинов – 145,
– крекинг – 185,
– коксование – 145 [50].
На рис. 12 приведены значения относительных скоростей реакций платформинга при температуре 500 С для различных парциальных давлений водорода.
Базовым уровнем является скорость дегидрирования, ее значение принято за 100 %.
Крекинг здесь представлен как сумма реакций гидрогенолиза и гидрокрекинга.
Для реакции образования кокса скорость при парциальном давлении водорода 10 бар принята за единицу.

Рис. 12. Скорость реакций платформинга
Дегидрирование нафтенов является самой быстрой реакцией платформинга, ее скорость в 7–8 раз превышает таковую для реакции изомеризации парафиновых и нафтеновых углеводородов и примерно в 30 раз скорость реакций крекинга и дегидроциклизации.
Реакция образования кокса является самой медленной реакцией платформинга.
Для реакций дегидроциклизации и крекинга константы скорости зависят также от длины углеродной цепи и увеличиваются при ее росте.
Особенно резкое увеличение констант скорости наблюдается для реакции дегидроциклизации при переходе от н-гексана к н-гептану, что объясняется статистическим фактором, а именно увеличением количества вариантов замыкания цепи.
Дегидроциклизация н-гексана в условиях бифункционального катализа протекает по схеме:
Лимитирующей стадией этих превращений является циклизация олефина с образованием 5-членного кольца.
Природа высокого энергетического барьера этой реакции может быть обусловлена циклической структурой активированного комплекса, являющегося переходным состоянием химической системы на ее пути от реагентов к продуктам реакции.
В соответствии с теорией активированного комплекса константа скорости реакции
где Δ H #и Δ S #– это изменение энтальпии и энтропии системы при образовании активированного комплекса.
Очевидно, что при образовании циклического комплекса энтропия системы уменьшается. Оценка изменения энтропии может быть сделана по изменению энтропии реакции циклизации.
Результаты представлены ниже в сравнении с изменением энтропии реакции изомеризации н-гексена-1 в 2-метилпентен-1 (табл. 4).
Таблица 4
Изменение термодинамических параметров при 800 К
Параметр
Изомеризация
С 5-циклизация
С 6-циклизация
Δ rG
Δ rH
Δ rS
–9500
+9400
+0,2
–15 600
–60 300
–56,0
–8300
–84 200
–94,8
Из данных табл. 4 следует, что при циклизации происходит значительное уменьшение энтропии. Применяя эти цифры для активированного комплекса, найдем отношение констант скорости реакции циклизации и изомеризации – 0,0012. Расчет отношения констант скоростей по энергиям активации дает такие же значение – 0,0012. Совпадение скорее случайное, но даже такой грубый расчет показывает, что вклад энтропии образования активированного комплекса может быть определяющим фактором низкой скорости С 5-циклизации.
Увеличение скорости циклизации при переходе от н-гексана к н-гептану приводит к существенному увеличению селективности ароматизации алкана.
В табл. 4 представлено также изменение энтропии реакции при С 6-циклизации 2-метилпентена-1: в этой реакции происходит еще более значительное уменьшение энтропии.
Если применить аналогичный подход для оценки отношения констант скорости двух альтернативных маршрутов циклизации, получим величину отношения С 6/С 5, равную 0,01. Это коррелирует с кинетическими данными, в соответствии с которыми С 5-циклизация является главным маршрутом дегидроциклизации парафиновых углеводородов риформинга на бифункциональном катализаторе.
Механизм циклизации достоверно не установлен.
В соответствии с гипотезой Гейтса [2], циклизация протекает по согласованному механизму с участием кислотного бренстедовского и основного льюисовского центров:
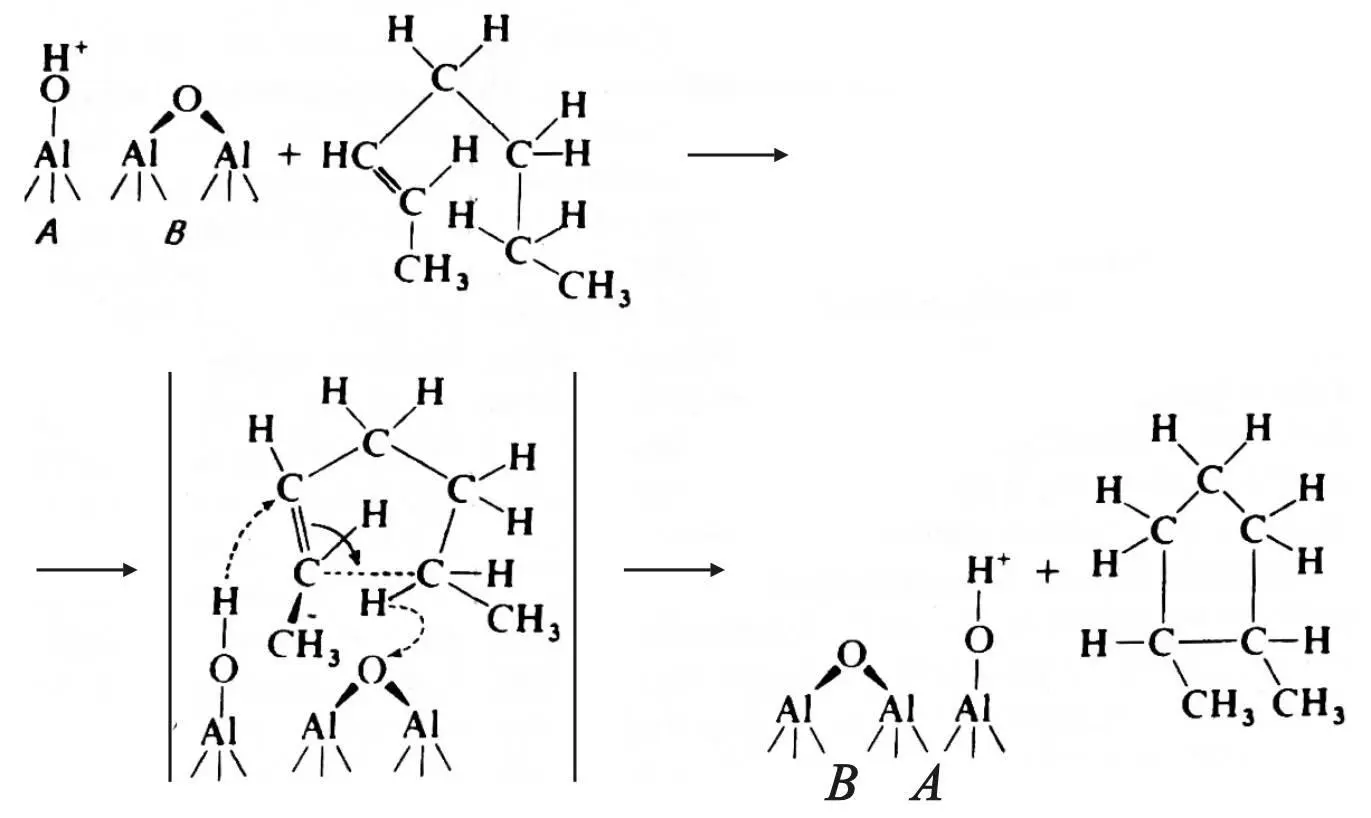
,
где А – кислотный бренстедовский центр; В – льюисовский основный центр.
Альтернативная гипотеза предполагает участие только льюисовских центров и поддерживается рядом экспериментальных фактов: отсутствие эффекта ингибирования азотом реакции циклизации, но ингибирование реакции расширения цикла. Известно также, что реакция циклизации может быть затруднена при увеличении влажности, что связывают с превращением льюисовских центров в бренстедовские.
При термодинамическом контроле направление и выход продуктов химического превращения определяются величиной и знаком изменения энергии Гиббса.
В случае кинетического контроля основным продуктом превращения является продукт реакции с меньшей энергией активации. Примером может служить превращение олефинов на кислотных центрах катализатора риформинга.
Из двух возможных химических реакций – изомеризации и гидрокрекинга – основным продуктом превращения является олефин, хотя его образование сопровождается меньшим понижением энергии химической системы. Превращение олефинов на кислотных центрах в условиях кинетического контроля обеспечивает высокую селективность процесса риформинга. Увеличение кислотности катализатора, повышение температуры процесса, увеличение времени контакта будет благоприятствовать протеканию реакции гидрокрекинга и снижению селективности ароматизации сырья.
Читать дальшеИнтервал:
Закладка:








